

理论
考虑溶液环境的分子动力学模拟,当离子距离生物分子足够近的时候,其实会导致生物分子的极化。而这一点,在传统的分子动力学力场中是没有考虑的。考虑了这一点而发展出的力场,被称作极化力场。
极化力场首先需要对生物分子进行重新建模。
对生物分子的所有非氢原子,额外绑定一个带电荷的虚原子,该虚原子与主原子之间以简谐势进行相互作用,简谐势的弹簧常数为
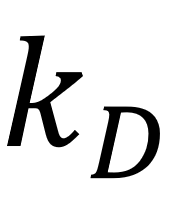 。总电荷
。总电荷 ,虚原子电荷
,虚原子电荷 ,主原子电荷
,主原子电荷 .
. 当没有外加电场的时候,虚原子以主原子
 为中心做简谐振动,整体仍表现为位置的点电荷。当外加电场
为中心做简谐振动,整体仍表现为位置的点电荷。当外加电场 时,虚原子将相对有一个位移
时,虚原子将相对有一个位移 ,该位移导致一个诱导的偶极矩
,该位移导致一个诱导的偶极矩 ,相当于该原子的极化率为
,相当于该原子的极化率为 。
。考虑该生物分子处于水溶液中,那么极化力场模型下,体系的总能量可以写成
 。
。其中
 ,
, 中没有Drude虚原子的贡献,后两项含有Drude虚原子的贡献,可以写成(s为溶剂分子)
中没有Drude虚原子的贡献,后两项含有Drude虚原子的贡献,可以写成(s为溶剂分子)
虚原子不参与LJ相互作用。
虚原子没有质量,但热运动还是有一份能量
 ,与质量无关。 这里的
,与质量无关。 这里的 可以保证虚原子满足热力学平衡,其值要远小于体系的
可以保证虚原子满足热力学平衡,其值要远小于体系的 。
。极化力场中,常用的水模型是SWM4-DP [1](还有一种SWM4-NDP[2])。NDP即negatively drude particle。考虑前面的偶极矩理论,drude粒子可以带正电,也可以带负电,但SWM4-DP用的更多一点。
操作
Lamoureux用的是CHARMM软件包。[1] CHARMM软件包非GPU版本在2015年之后开源了,但效率比较低。
而NAMD与CHARMM有着比较好的配合,二者都是psf文件存储结构信息。力场文件也比较相似,NAMD 2.12版本之后,可以直接读取CHARMM力场的str文件。但需要2.14版本,才对Drude有更好的支持。(下面的例子用的也是NAMD 2.14版本)。
NAMD的Drude力场官方教程基于的是2011年的J. Phys. Chem. Lett.文章。[3]
官方教程里给出了三个例子,water, decane, NMA。这些文件其实都是charmm软件包生成的。
对于decane的例子,给出的是真空情况,如果要solvate,在VMD里直接使用solvate plugin是不行的,因为solvate plugin默认是没有SWM4水模型的。
对于生物分子的水溶液体系,目前建议使用CHARMM-GUI网站的Drude Prepper工具生成。[4] 步骤是先生成常规力场下的水溶液体系(可以包含离子),将psf文件和pdb文件上传之后,Drude Prepper工具会自动转换成Drude力场下的模拟初始文件,除了适用于CHARMM软件包的初始文件,还可以选择NAMD软件包的初始文件,非常方便。
特别是,还提供了生成最终模拟文件的CHARMM软件配置文件。即,基于给出的配置文件,使用本地的CHARMM软件,自己也可以得到Drude力场的模拟初始文件。
但即使只需要上传psf文件和pdb文件,当体系比较复杂的时候,也会出现各种问题,导致Drude Prepper工具报错。这个时候,懂一点CHARMM的语法,是很有帮助的。
下面我就以namd教程里的1UBQ.pdb为例[所有文件:drude-1ubq.zip],说明如何基于CHARMM来得到1UBQ蛋白在SWM4水溶液中的Drude极化力场初始模拟文件。其中CHARMM的代码文件参考了CHARMM-GUI网站里的工具。
首先从1UBQ.pdb开始。
根据1.tcl,可以得到ubqp.pdb,这步是为了去掉结构里的水分子。
可以采用vmd非界面模式:vmd –dispdev text –e 1.tcl
再根据2.tcl,可以得到ubq.pdb, ubq.psf,2.tcl里将HIS命名为HSE,不然charmm不清楚HIS的质子化状态.
ubq.pdb就可以作为charmm的1.inp的输入文件了。1.inp读取charmm力场,对初始pdb文件进行处理,得到Charmm格式(crd)的坐标文件。
再根据2.inp文件,生成指定大小(需要修改2.inp的指定行)的水盒子 (step2.1_waterbox.crd,文件等)。
再根据3.inp文件,得到加了指定浓度离子(可以在step2.2_ions.prm里指定,但step2.2_ions.crd需要对应修改)的1ubq水溶液体系。(step2_solvator.psf和step2_solvator.crd等)。
如果不进行Drude力场计算,这些文件是可以作为常规力场的初始文件的。
要进行Drude力场计算,需要进行如下几步转换:
根据4.inp,该文件读取toppar_drude文件下的Drude力场文件,对蛋白、水分子和离子的模型进行替换。每种模型被分别处理成一个segment,得到step1_reader_seg1.crd, step1_reader_seg2.crd, step1_reader_seg3.crd, step1_reader_seg4.crd等文件。
再由5.inp, 配合crystal_image.str,checkfft.py和toppar_drude.str,得到Drude力场下体系的psf文件。(step2_drude.psf等)。
注意5.inp中, Setup crystal处的参数要设定为盒子的大小。
然后6.inp, 对所得到的体系进行能量最小化,得到文件step2_drude.pdb, step2_drude.crd和文件夹restraints。
到此为止,step2_drude.pdb和step2_drude.psf便可以用于NAMD计算了。
使用1.namd, 进行em+nvt.
如果报错,一般是em的步骤不够多。
刚开始nvt步骤,最好跟再em步骤之后,放到一个namd文件里。
这里的案例,10000 step em是足够的。
1. Lamoureux, G., A.D.M. Jr., and B.t. Roux, A simple polarizable model of water based on classical Drude oscillators. The Journal of Chemical Physics, 2003. 119(10): p. 5185-5197.
2. Lamoureux, G., et al., A polarizable model of water for molecular dynamics simulations of biomolecules. Chemical Physics Letters, 2006. 418(1): p. 245-249.
3. Jiang, W., et al., High-Performance Scalable Molecular Dynamics Simulations of a Polarizable Force Field Based on Classical Drude Oscillators in NAMD. The Journal of Physical Chemistry Letters, 2011. 2(2): p. 87-92.
4. Jo, S., et al., CHARMM-GUI: A web-based graphical user interface for CHARMM. Journal of Computational Chemistry, 2008. 29(11): p. 1859-1865.
转载本文请联系原作者获取授权,同时请注明本文来自宋永顺科学网博客。
链接地址:https://m.sciencenet.cn/blog-485752-1291836.html?mobile=1
收藏