博文
量子编码与化学
||
量子编码与化学
左芬
上海微观纪元数字科技有限公司
近日,我们团队利用量子编码技术揭示出化学体系中简单而优美的量子纠缠结构,并在此基础上快速求解出体系基态的近似能量。这一突破性进展有望在计算化学和量子算法领域均引起重大变革。
量子力学的建立为化学体系中电子结构的描述提供了基本原理,也就是Schrödinge方程。可是,Schrödinger方程的精确求解是极为困难的。对于只有一个电子的氢原子和类氢离子,我们可以得到解析解。可是对于哪怕只有两个电子的H2分子,我们到现在为止仍然只能依赖极为繁琐的数值方法。无奈之下,物理学家/化学家们想出了一套近似求解方法:Hatree-Fock近似。简单来说,就是把电子一个个隔离出来单独求解,再以适当的方式拼在一起。如果电子之间相对独立,这么做的结果还不错;但如果电子之间存在很强的关联,或者说量子纠缠,其结果往往很差。如下图所示,Hartree-Fock近似给出的H2能量在键长较小时与精确值较为接近,但在键长较大时相去甚远。。后来人们又在Hatree-Fock近似基础上做了各种改进,但并没能从根本上解决这一问题。
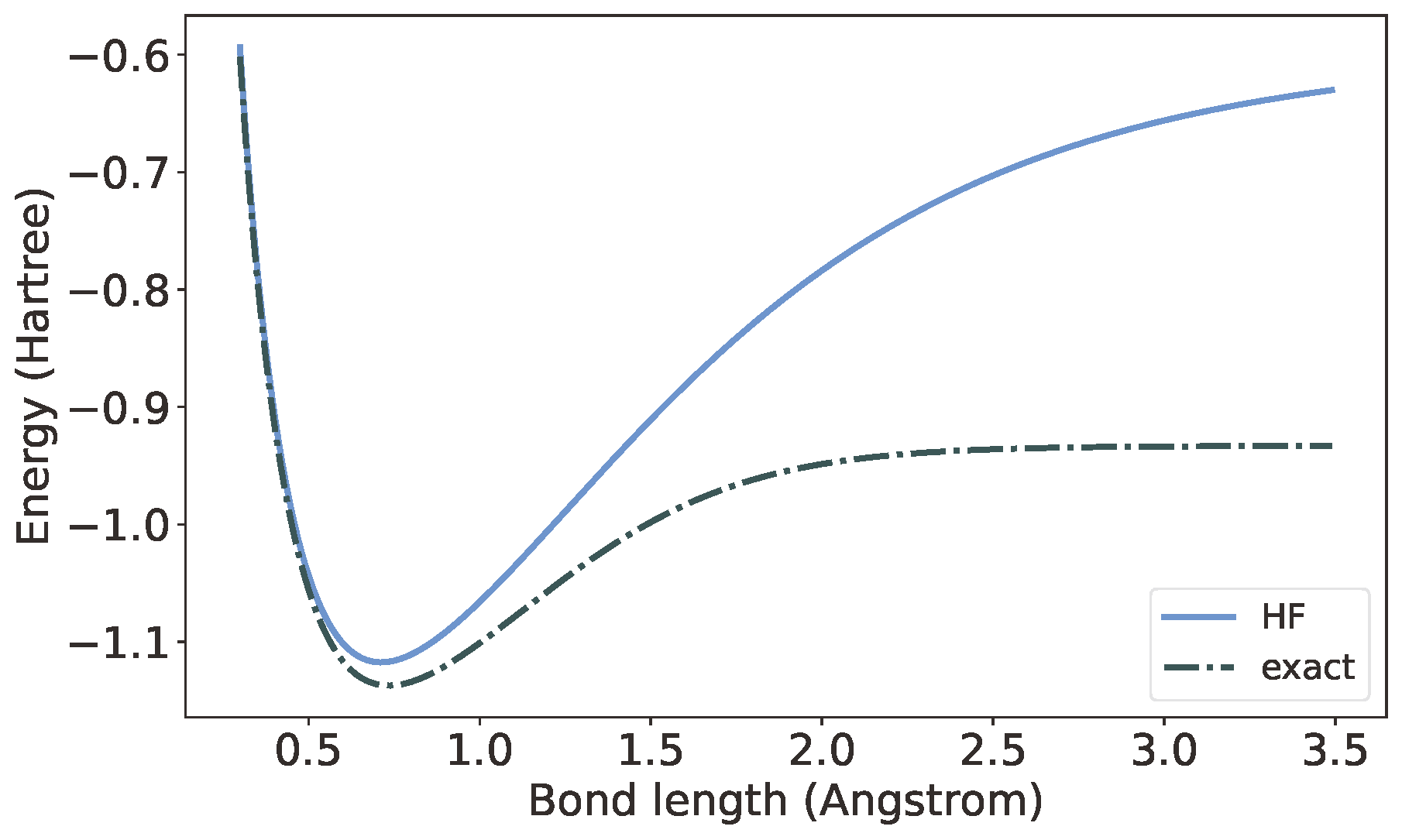
有一种观点认为,电子的费米子特性是这一难题的根源。费米子的诡异性质之一就是,交换两个相同的费米子会带来一个负号,要再交换一次才能还原。与之相对的,跟我们的直觉更相符的粒子则被称为玻色子,例如光子。早在1928年,Jordan和 Wigner就找到了将有序费米子体系转换为玻色子体系的方法。量子计算兴起之后,因为实际构建的量子比特都是玻色子,人们又发明了另外两种将费米子转换为玻色子的变换方法。可是,做了这些变换之后,电子相互作用的Hamilton量(可以简单地理解为能量)的表达式似乎更加复杂了,相应的Schrödinger方程也更加复杂。这就使得寻找电子纠缠结构的问题更加扑朔迷离。
我们团队得以初步解决这一难题的关键是利用了量子信息学家们在上世纪末发展起来的量子编码技术,特别是其中的稳定子(stabilizer)表述。对于物理学家来说,有一个非常直接的方法来理解这一表述:所谓稳定子,可以粗略地理解为“一组相互对易的力学量”;而稳定子态,就是它们的“共同本征态”,且本征值均为1。只不过对于量子比特来说,稳定子表述具有更加丰富而优美的数学结构。有了稳定子这一“火眼金睛”,电子体系的纠缠结构可以说一目了然。下面我们仍然用H2分子的基态来简单说明。
我们知道H2分子有两个原子轨道,考虑到电子自旋,会有四个自旋轨道。相应地,我们可以用四个量子比特去描述。利用对称性可以将量子比特数削减(tapering)为两个。这一步不是必需的,但可以让结果更加显然。这样一来,我们会得到如下的Hamilton量(键长为0.74Å):
H = -1.0534210769165204 * II
+ 0.39484436335590356 * IZ
- 0.39484436335590367 * ZI
+ 0.1812104620151969 * XX
- 0.011246157150821112 * ZZ.
其中I是2*2单位矩阵,X,Y,Z是Pauli矩阵。当键长很大,比如在2.8 Å时,Hamilton量如下:
H= -0.8284676561247681 * II
+ 0.016170000066607376 * IZ
- 0.016170000066607328 * ZI
+ 0.2930431286727852 * XX
- 0.0001469354633982234 * ZZ.
请一定要注意系数大小的变化! 现在我要告诉你,对于前者,稳定子是-IZ和ZI,稳定子态是|01>,也就是直积态/Hatree-Fock态; 对于后者,稳定子是-XX和ZZ,稳定子态是两比特纠缠态:
.
如果你没能一眼看出来,说明你还需要回太上老君的八卦炉里再修炼一阵。当然也许这个问题并不像我说的这么简单。因为据说化学家们20多年前就已经得出Hamilton量的类似表达式了,但他们似乎并没能从中看出电子的纠缠行为。
将每个距离处的Hamilton量分别作用到这两个态上,再取它们中的较小值,会得到以下曲线:

可以看到,仅仅利用一个简单的两比特纠缠态,就几乎找回了全部的电子关联能!让人不得不感叹,原来真的是“大道至简”!
你可能会怀疑这是因为H2分子比较简单,才会有这么简单的纠缠行为。我们进一步在 LiH和BeH2体系中做了探索,发现了极为相似的纠缠结构。简单来说,就是这些分子会先把多出的电子按照Hartree-Fock方法填到轨道中,再把最后的两个活跃电子按照与H2类似的方式纠缠在一起。我们下一步打算研究更加复杂的分子,以求找出更加复杂的纠缠形式。
你可能还会问,你利用纠缠结构并没能得到图中的精确基态能量啊。的确如此。量子纠缠只是量子计算的骨架,还需要一些更加精细的操作才能让她血肉丰满起来,进而发挥全部威力。大自然也同样如此。
最后我们借用著名华人物理学家文小刚先生的一些理念来作为总结。他认为,大自然在根本上其实是一些相互纠缠的量子比特的海洋,而所谓的费米子特性不过这些量子比特集体运动呈现出的一种表面现象。在这里的化学体系中,大自然先把量子比特两两纠缠起来,再把它们伪装成费米子,用这样的双重编码蒙蔽了我们近一个世纪。如今我们利用量子计算和量子信息技术,终于开始逐渐揭开大自然的神秘面纱,这怎不令人激动万分?
论文信息:
Stabilizer Approximation, Xinying Li, Jianan Wang, Chuixiong Wu, Fen Zuo, https://arxiv.org/abs/2209.09564。
https://m.sciencenet.cn/blog-863936-1356472.html
上一篇:量子信息的物理
下一篇:量子纠缠与最小团覆盖
全部作者的精选博文
全部作者的其他最新博文
- • 随机性的统一理论
- • 布朗地图
- • 迷宫证明为统计力学塑造“脊梁”
- • Ising模型是NP-完全的
- • Ising模型众家谈
- • 退量子化