博文
sam/bam格式解读之Edit Distance编辑距离(NM tag)
|
sam文档中对NM tag (编辑距离: 从字符串a变到字符串b,所需要的最少的操作步骤(插入,删除,更改)为两个字符串之间的编辑距离。)
举个栗子:两个字符串“eeba”和“abca”的编辑距离是多少?
根据定义,通过三个步骤:1.将e变为a 2.删除e 3.添加c,我们可以将“eeba”变为“abca”。

所以,“eeba”和“abca”之间的编辑距离为3。

回到序列比对的问题上:
下面是常见的二代比对到ref的结果(bwa):
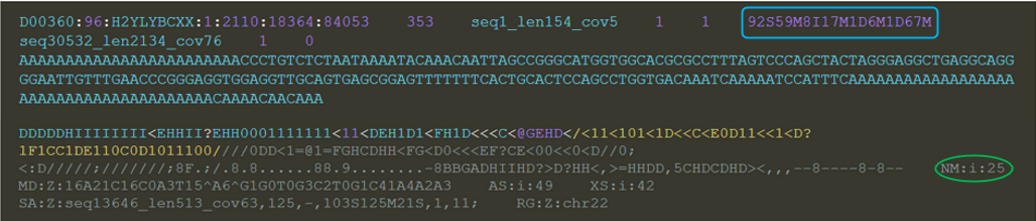
ref序列:

【说明】蓝框为cigar字段,值+比对标志符;绿框为NM tag值
Query: 249bp | Sbjct: 154bp
CIGAR string: 92S 59M 8I 17M 1D 6M 1D 67M(对象是Query,即确定Query每个碱基比对参考序列的情况)
如下图解释:
92个被剪切(一般在两端出现),8个插入,17个匹配和错配,1个缺失,6个匹配和错配,1个缺失,67个匹配和错配

根据cigar字段可以统计indel信息,但是无法统计mismatch(错配,上图红色箭头)。
公式:mismatch = NM - I - D = 25 – 8 – 1 – 1 = 15
【参考】
cigar字段包含了序列比对的简化信息 | |||
M | match and mismatch | 匹配和错配 | 常用 |
I | insert | 插入 | |
D | deletion | 删除 | |
N | skipped | 跳过该区域,表示可变剪接位置 | 不常用
|
S | soft clipping | 被剪切的序列存在与序列中 | |
H | hard clipping | 被剪切的序列不存在与序列中 | |
P | padding | 缺口 | |
= | match | 纯match | 少见 |
X | mismatch | 纯mismatch | |
NM | edit distance | NM tag | sam/bam |
M/I/S/=/X:这些数值的加和等于sam文件第10列SEQ的長度
【备注】clipped均表示一条read的序列被分开,之所以被分开,是因為read的一部分序列能匹配到第三列的RNAME序列上,而被分开的那部分不能匹配到RNAME序列上。H只出現在一条read的前端或末端,但不會出現在中间。S可以单独出現,而H必須有与之对应的S出现時才可能出现。
| soft clipping:是指虽然比对不到基因组,但是还是存在于SEQ (segment SEQuence)中的序列,此时CIGAR列对应的S(Soft)的符号 |
| hard clipping:表示比对不上并且不会存在于SAM/BAM文件中的序列(被截断扔掉了的序列,此时CIGAR列会留下H(Hard)的符号,但是序列的那一列却没有对应的序列了) |
10H10M10H:那么seq的长度是10bp 10S10M10S:那么seq的长度是30bp |

Edit Distance编辑距离(NM tag)- sam/bam格式解读进阶
https://m.sciencenet.cn/blog-994715-1362775.html
上一篇:QIIME 2序列处理情况汇总详解
下一篇:Git简明使用教程