博文
非化学计量比材料设计:虚晶近似、高通量计算、机器学习
|||
关注:非化学计量比材料设计:
1) 虚晶近似、Matcloud、高通量计算、机器学习、结构预测
2) 相图构建
3) 离子型化合物和金属型化合物稳定性判据
https://www.asminternational.org/phase-diagrams
金属活动性越强,就是还原性越强,越容易被氧化,所以是越不稳定。
https://baike.baidu.com/item/%E9%87%91%E5%B1%9E%E6%80%A7/1775204?fr=aladdin
举个例子吧。氢→钠→钾→铷→铯→钫 的金属性是逐渐增强的,它们的热稳定性却是越来越低,它们的氢化物稳定性也是越来越低。
�
https://wenku.baidu.com/view/6abe8998ff00bed5b9f31de2.html
单质越易跟H2化合,生成的氢化物也就越稳定,氢化物的还原性也就越弱,其非金属性也就越强。
�
金属性是指在化学反应中金属元素失去电子的能力。失电子能力越强的粒子所属的元素金属性就越强;反之越弱,而其非金属性就越强。金属性常表示元素的原子失去电子的倾向;元素的非金属性是指元素的原子得电子的能力。
一般说来,元素的金属性越强,它的单质与水或酸反应越剧烈,对应的碱的碱性也越强。例如:金属性Na>Mg>Al,常温时单质Na与水能剧烈反应,单质Mg与水能缓慢地进行反应,而单质Al与水在常温时很难进行反应,它们对应的氧化物的水化物的碱性 NaOH>Mg(OH)2>Al(OH)3。
元素的非金属性越强,它的单质与H2反应越剧烈,得到的气态氢化物的稳定性越强,
元素的最高价氧化物所对应的水化物的酸也越强。例如:非金属Cl>S>P>Si,Cl2与H2在光照或点燃时就可能发生爆炸而化合,S与H2须加热才能化合,而Si与H2须在高温下才能化合并且SiH4极不稳定;氢化物的稳定HCl>H2S>PH3>SiH4;这些元素的最高价氧化物的水化物的酸性HClO4>H2SO4>H3PO4>H4SiO4。因此,在化学反应中的表现可以作为判断元素的金属性或非金属强弱的依据。
非金属性越强,该元素电负性越强,和氢原子生成的共价键越强,对应的氢化物的稳定性越强.
有两种氢化物,金属的氢化物属于离子化合物,非金属的氢化物属于共价化合物,下面判断氢化物稳定性。
金属元素越活泼,半径越小,氢化物越稳定,如NaH>MgH2,LiH>KH。
金属氢化物稳定性难总结规律,也不是中学考点。
非金属元素的非金属性越强,对应气态氢化物的稳定性也就越强,在非金属中:
①同一主族中,表现为由上往下气态氢化物的稳定性递减 。
②同一周期中,表现为从左往右气态氢化物的稳定性递增 。
成键原子之间的键能越大,气态氢化物的稳定性也就越强.
结构决定性质,离子键的强弱与离子化合物的稳定性有关,准确的说应该是离子键的强弱决定了离子化合物的稳定性。
应该反过来说:离子化合物的稳定性与离子键的强弱有关,离子键越强离子化合物越稳定。而阴阳离子的相互作用力越大离子键越强;阴阳离子间的作用力与离子的电荷数及间距有关
vegar's law:
http://blog.sciencenet.cn/blog-2321-738776.html
Vegard’s Law可能是唯一被写入教科书的,并且在多数情况下不成立的物理定律。由此可见,Vegard定律所表述的深刻内涵是何等的重要啊!
Vegard定律简单地给出固溶体晶格常数随组元成分变化的线性关系式,在多数情况下,Vegard定律与实验事实不符,但是人们习惯上却说实验偏离了Vegard定律。
经过10多年的思考,我看到了Vegard’s Law背后的物理问题,即原子和原子究竟如何接触。因此,我对Vegard敬佩有加,因为他于1921年为我提出了一个有相当含金量的科学问题。至于实验结果是否与他定律的计算结果相符合,这只是两个数据的对比,差别背后隐藏着的秘密,才是我梦寐以求的东西。
1921年,挪威物理学家Lars Vegard (1880-1963)发表了一篇文章(Z. Phys. 5 (1921) 17),提出了一个非常简单的公式(我用连续固溶体表述):
a=x1a1+x2a2 (1)
式中,a1和a2分别为组元1和组元2的晶格常数,x1和x2分别为组元1和组元2的百分含量,a为固溶体的晶格常数。
就这么一个简单的公式,而且在绝大多数情况下和实验结果并不相符,即计算结果和实验结果相比是很离谱的。但是,Vegard’s Law却出现在越来越多的现代科学文献中。
Deviationfrom Vegard’s law(Google搜索)
Departure from Vegard’s law(Google搜索)
据Wikipedia介绍,Vegard除了有个著名的Vegard’s Law之外,映像到现在的几乎一无所有(还研究过北极光)。(欢迎网友提供更多Lars Vegard的信息)
每当我思考Vegard’s Law的时候,我就会想到这样的问题:
与其发表几百篇SCI文章,还不如搞一个不太好用的定律,例如,Vegard’s Law。
从1993年起,我全方位(以后展开讲)地对Vegard定律思考了20多年,得到了别人得不到或没有得到的东西。换句话说,我从Vegard定律中,挖掘到了我想要的东西,其中最重要的idea就是原子和原子究竟如何接触?
Deviationfrom Vegard’s law(Google搜索)
Departure from Vegard’s law(Google搜索)
实验偏离了Vegard定律,这种表达方式,不仅仅是个语言问题,还有更深刻的物理背景蕴含其中(Vegard提出了一个科学问题)。
20年来我异想天开地思考Vegard定律,我得到了什么呢?请听我慢慢道来!
让我们先回到教科书,看看现在的权威教科书对Vegard’s Law的描述:

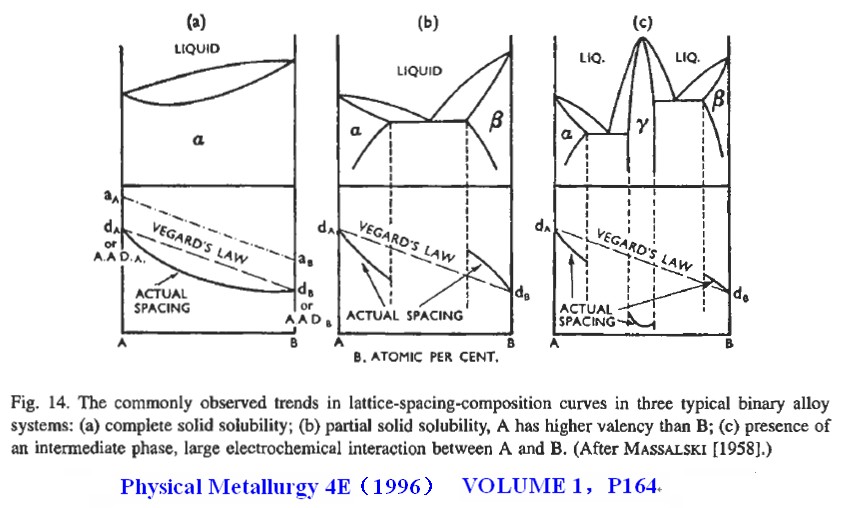
在多数情况下,Vegard定律与实验事实不符,但是人们习惯上却说实验偏离了Vegard定律。
我感兴趣的问题是:
实验为什么偏离了Vegard定律?
要回答这个问题,还需要从原子的模型讲起。
如果原子是刚性的(刚球模型),那么,对于连续固溶体体系,Vegard定律就不会偏离实验结果。换句话说,组元1和组元1混合形成固溶体时,如果原子是刚性的,那么,固溶体的晶格常数应该就是a=x1a1+x2a2。
问题是绝大多数实验结果和Vegard定律不符,因此,可以得到推论1。
推论1:
原子不是刚性的。
以Cu-Ni连续固溶体为例,进一步表述如下:
当组元1(Cu)和组元2(Ni)混合形成固溶体时,由于原子不是刚性的,组元Cu原子要收缩,而组元Ni原子要膨胀,如果Cu原子的收缩量和Ni原子的膨胀量相等,那么,固溶体的晶格常数仍然应该就是a=x1a1+x2a2。
问题是绝大多数实验结果和Vegard定律不符,因此,又可以得到推论2。
推论2:
是Cu原子的收缩量和Ni原子的膨胀量不相等,这才导致了固溶体的晶格常数不能再用a=x1a1+x2a2描述。
推论3:
如果Cu原子的收缩量大于Ni原子的膨胀量,那么,和Vegard定律的直线比,固溶体的晶格常数发生了负偏离;
推论4:
如果Cu原子的收缩量小于Ni原子的膨胀量,那么,和Vegard定律的直线比,固溶体的晶格常数发生了正偏离。
(根据原子界面处电子密度连续,可知Cu原子收缩,Ni原子膨胀)
推论3或推论4是否正确,最后要用实验数据来裁决!
以上推论3和推论4,是我对Vegard定律思考多年得到的深刻体会,即这就是隐藏在Vegard定律背后的秘密,我找到了。^_^
Vegard定律只是我思维的一个载体,于是,我先把Vegard定律背后的秘密发表,然后再回头写一篇关于Vegard定律的原子模型(已经被欧洲某物理刊物接受)。
TFDC相图(2003年,自然科学进展,点击可下载)
Atomic Phase Diagram(2004年,Progress in Natural Science,点击可下载)
英文版文章里提到:The atomic model of Vegard’s Law will be published.
中文版文章里也提到:《Vegard定律的原子模型》将另文发表;
为什么《Vegard定律的原子模型》10年后(2003年到2013年)才发表出来?
这是另外一个故事了,还是等文章出来之后再讲述吧!
Atomic Phase Diagram被别人实质性地引用了一次(引用一个图)。
Hardness of metallic crystals(在正文里引用了我的一个图^_^)如果仅仅是在Introduction里引用了我的文章,那就不值得一提了。 我的“原子相图”可以用来组装原子,《Vegard定律的原子模型》算是一个简单应用的铺垫例子。还需要另外一个不同应用的铺垫例子。
“昨夜西风凋碧树, 独上高楼, 望尽天涯路”。
在我的脑子里,已经有了模糊的物理图像,但是,要把这个模糊的物理图像清楚地表达出来,可能需要好几年的时间。就像对Vegard定律的思考一样。
不过,我已经做好了准备!
虚晶近似
http://blog.sciencenet.cn/blog-749450-775494.html
An approach is to use the so-called “virtual crystal approximation” (VCA).The virtual crystal approximation is based on building a fictitious “virtual atom” potential by averaging the ionic potentials of the atoms that alternate at the same position in the structure [17].The VCA method has been proven to successfully investigate the alloys containing not only monovalent atoms but also heterovalent atoms [18-20].Given the mixed pseudopotentials have similarities, it seems to be able to play a role to a certain extent. In this paper, VCA calculations are employed to investigate structural, electronic, dynamic and thermodynamic properties of Zr1-xHfxH2. We utilized the VCA where the alloy pseudopotential was constructed within the first-principles VCA scheme for the handling of the disordered ternary alloy. Elemental ionic pseudopotentials of ZrH2 and HfH2 are combined as:
![]() (1)
(1)
to construct the virtual pseudopotential of Zr1-xHfxH2.
听人说abinit可以计算虚晶近似,居然不知道何谓虚晶近似,更谈不上进行计算,现求助各位高手,能否分享一下资料。
李正中 固体理论12章 平均t矩阵
一个原子,看成两种或以上种原子的混合,用百分量表示含量。
使用该方法请谨慎,原子混合后计算结果正确性难以保证!
虚晶近似一般是用来做参杂体系的.由于要模拟真实的参杂体系需要考虑很多原子,因此才引入了虚晶.比如要做金刚石10%掺B的模拟,可以在C的赝势中考虑3.6个电子.但由于把参杂的效应给平均化了,一些参杂导致的局域化是模拟不出来的.
确实看到很多人都对VCA的结果表示怀疑,可是现在对这种低含量掺杂还有其他更好的建模方法么?望指教!
可以建立大超胞做Quasi-Random的模型,只不过计算量就有点过份了,没有大型机搞不定的~
:D 那么,是哪个软件支持呢?简单的VCA的话,CASTEP和PWscf都支持的~
FPLO有,还有AKAI-KKR和SPRKKR,但是那些处理金属比较好,处理绝缘体或者半导体效果不太好。
个人感觉是不靠谱的 只是一种算法,背景如下
VCA就是virtual crystal approximation,比如对于无序系统,Ca(Fe0.5Nb0.5)O3,Fe和Nb同时占据复合钙钛矿结构的B位的时候,无无序系统传统的从头算方法貌似无法解决,如果要计算就只能假定一比一有序,用有序计算的结果模拟无序体系,这有两个劣势:一个是假定一比一有序可能要用超晶胞,增加了计算量,因为平面波软件计算量是随着原子个数(胞体积)三次方增加的;另一个劣势是假定一比一有序可能不足以模拟无序系统。
VCA方法是可以指定Fe和Nb原子同时占据钙钛矿B位,计算的时候根据Fe和Nb原子的原子序数,核电荷数,质量,电子数,根据Fe0.5,Nb0.5的计量比,构造一个赝原子,也就是假的原子,在B位,简单说也就是平均了Fe和Nb的贡献,构造一个假的原子。这种做法感觉不靠谱,不过也没试过。
castep是支持的,另外ABINIT也是支持的,abinit如果你想试试,可以在abinit的input中找mixach这个关键词(也可能是mixalch,记不清了),之后按照这个关键词的相关链接,即可找出VCA方法的关键词
顺便补充一下,PWscf也可以通过upftools/virtual.x对赝势进行混合实现VCA方法。不过混合时对赝势的要求比较高,要求建立赝势时的很多参数必须能够相互对应。所以从PWscf官方提供的赝势库中,能制作混合赝势的并不多……
另外,90年代时,Baroni等人似乎也用VCA方法进行DFPT计算来着(不知道Abinit可以不可以,反正CASTEP不能在VCA下再计算声子),事实上,如果能在PWscf中得到混合赝势,的确是允许DFPT计算的。
顺便给LSS论文,这是我第一次看到Quansi-Random模型,至于还有没有其他的类似的方法或者这个方法的起源什么的,我就没再追究了,因为本人做计算都是用自己的电脑,比较悲催,大点的模型都算不太动……
A. Chroneos, C. Jiang, et. al, Defect interactions in Sn(1-x)Ge(x) random alloys, Applied Physics Letters, 94, 252104, 2009.
https://m.sciencenet.cn/blog-567091-1126202.html
上一篇:再关注:Raman光谱、超导温度计算、U值选择、HSE或W、力学常数计算
下一篇:等离子体放电再谈:射频vs微波