博文
转录组数据的分析
||||
转录组数据的分析
(1)转录组测序,以两组比较实验为例,实验组为T,对照组为C,每组三个重复,T1,T2,T3,C1,C2,C3。诺禾致源返回的文件为T1_1_clean.fq,T1_2_clean.fq,T2_1_clean.fq,T2_2_clean.fq,T3_1_clean.fq,T3_2_clean.fq,C1_1_clean.fq,C1_2_clean.fq,C2_1_clean.fq,C2_2_clean.fq,C3_1_clean.fq,C3_2_clean.fq。需要注意:如果测序结果中含有接头序列,需要剔除。
(2)分析方法参考文献”Differential gene and transcript expression analysisssss of RNA-seq experiments with TopHat and Cufflinks“。
(3)操作系统:乌班图优麒麟
(4)软件安装:在/home/usrname/文件夹(也就是~/)下建立bin文件夹:#mkdir /home/usrname/bin。将这个文件夹添加到系统路径中# export PATH=$PATH:/home/usrname/bin (如不了解此命令,可以到网上查,不要把默认的系统路径删除)。
(5)下载samtools(samtools-0.1.17.tar.bz2,版本可以改变,但我这里没有试过,过高的版本需要配置);bowtie(bowtie-0.12.7的linux的二进制版本,太高版本需配置参数);tophat(1.3.2的linux的二进制版本)。解压了bowtie和tophat压缩包后,里面的二进制文件全部拷贝到/home/usrname/bin下。解压samtools后,进入文件夹,# make,然后文件夹里生成可执行文件samtools,将之拷贝到/home/usrname/bin下。
(6)在~文件夹下新建文件夹,比如“RNAseq_XXX”,将诺禾致源返回的clean.fq文件拷贝进去(如果为fq.gz,则# gunzip ./*,将之解压)。
(7)建立基因组索引文件,并下载GTF(General/Gene transfer format)文件。首先,下载基因组序列(注意基因组的版本号与注释文件的版本号要一致)。比如,下载UCSC hg38基因组http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz。可以将基因组文件合并,用cat *.fa > XX.fa,也可以不合并。基因组文件下载完毕之后,# bowtie-build 基因组.fa 基因组名(比如hg19.fa,可以写hg19,此是bowtie生成的基因组文件的前缀)。
GTF文件可以用如下方式下载:
http://genome.ucsc.edu/cgi-bin/hgTables,
�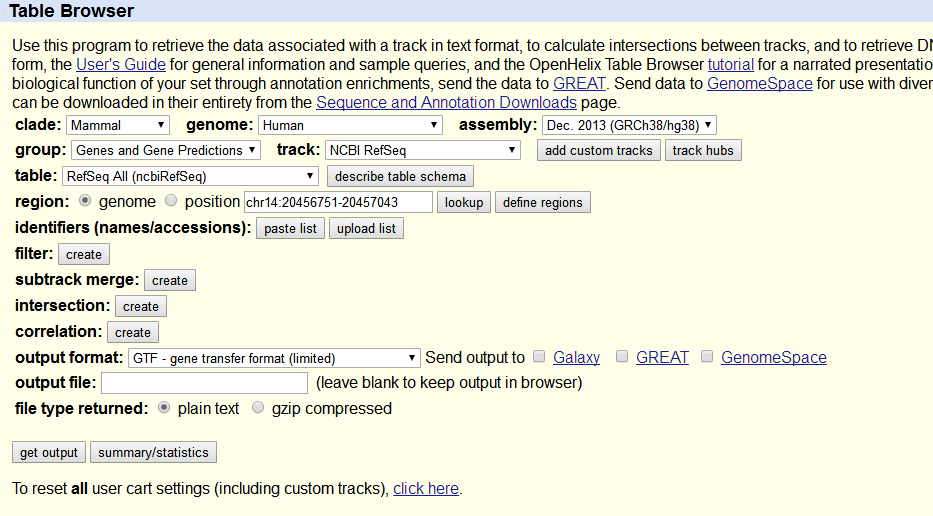 �
�
点击get output。
(8)比对(此步骤耗费很长时间,可多线程并行,但不解决速度问题。我的每个重复耗时一天,则大约一周运行完。)。建立符号链接#ln -s GTF_file_path workingDirectoryPath,#ln -s Bowtie_index_filePath/* workingDirectoryPath。如果不创建符号链接,可能会报错。
# tophat -p 8 -G GTF文件路径 -o TH_输出文件的文件夹 基因组索引位置(所在文件夹/索引前缀) fq文件(可以多个,中间以空格分开)。
这一步运行完之后,会生成日志文件夹、accepted_hits.bam文件、deletions.bed文件、insertions.bed文件、junctions.bed文件、left_kept_reads.info和right_kept_reads.info文件。
(9)运行完以上程序,或每个重复运行完后,执行如下命令:
# culllinks -p 8 -o CL_输出文件夹 TH_输出文件的文件夹/accepted_hits.bam
这步运行完后,会生成四个文件:genes.fpkm_tracking,isoforms.pfkm_tracking,skipped.gtf,transcripts.gtf。
(10)整合转录组注释。
先构建一个文档文件,名为assemblies.txt,内容如下
./CL_输出文件夹1/transcripts.gtf
./CL_输出文件夹2/transcripts.gtf
./CL_输出文件夹3/transcripts.gtf
./CL_输出文件夹4/transcripts.gtf
./CL_输出文件夹5/transcripts.gtf
./CL_输出文件夹6/transcripts.gtf
然后
# cuffmerge -g GTF文件路径 -s 基因组.fa 8 assemblies.txt
漫长的等待后(一个小时)
(11)鉴定差异表达基因。
# cuffdiff -o diff_out -b 基因组.fa -p 8 -L group1Name,group2Name(注意逗号) -u merged_asm/merged.gtf ./TH_输出文件的文件夹1accepted_hits.bam,./TH_输出文件的文件夹2accepted_hits.bam,./TH_输出文件的文件夹3ccepted_hits.bam(注意粉色逗号) (此处有空格,前边是第一组的bam文件,后边是第二组的bam文件)./TH_输出文件的文件夹1accepted_hits.bam,./TH_输出文件的文件夹2accepted_hits.bam,./TH_输出文件的文件夹3ccepted_hits.bam
漫长的等待后(大概24小时),将diff_out文件夹拷贝到windows系统,分析结果。
https://m.sciencenet.cn/blog-331314-1133590.html
上一篇:来MRC LMB工作的感受
下一篇:乌班图操作系统与软件的安装