博文
内部翻译资料:泛素与苏木在DNA损伤响应中发挥的调控作用
||||
《分子细胞》杂志综述
泛素与苏木在DNA损伤响应中发挥的调控作用 (Regulationof DNA Damage Responses by Ubiquitin and SUMO)
作者:英国剑桥剑桥大学戈尔登研究所与生物化学部,杰克逊史蒂芬
加拿大多伦多西奈山医院伦恩菲尔德塞缪尔研究所,多伦多大学分子遗传部,堵罗赫丹尼尔
通讯邮箱:s.jackson@gurdon.cam.ac.uk (S.P.J.),durocher@lunenfeld.ca (D.D.)
摘要:
以泛素与苏木多肽共价结合靶蛋白著称的泛素化与苏木化,是细胞功能控制中普遍存在的机制。在此,我们总结了,在泛素与苏木共轭结合底物过程中的关键步骤和关键酶,并概述它们何以在维持基因组稳定性方面发挥紧要的作用。特别的,我们综述了泛素化与苏木化在DNA损伤识别、送信和修复的生化、细胞与整个机体层面发挥的调控与协调机制。此综述除了对DNA损伤与相关通路的调控和重要性做深入剖析外,由此建立的范式也可普及到其它的细胞进程,或可为更好理解与治疗人类疾病提供裨益。
泛素化与苏木化之基本原理
泛素化者,是将76氨基酸长的残余蛋白泛素共价结合到其它蛋白上的过程,起初被认为是靶向目的蛋白,并转交蛋白酶体毁解的机制,现在则认为它还参与蛋白的活性、定位于互作(Berginkand Jentsch, 2009; Komander and Rape, 2012)。除了泛素,细胞内还存在甚多与泛素蛋白结构相关的类泛素蛋白(ubiquitin-likeproteins,UBL)。泛素和多数UBL都是依赖E1,E2,和E3连接酶将其碳末端的甘氨酸残基连接到靶蛋白上的(图一)。UBL中被广泛研究的是大约100个氨基酸长短的苏木蛋白(小泛素相关调节子)。真核生物通常是多个基因表达一种类型的泛素。但是脊椎动物的苏木却有两种:SUMO-1和高相关度且功能允余的SUMO-2与SUMO-3(SUMO2/3)。低等生物里,例如酿酒酵母和裂殖酵母,它们中只存在一种苏木蛋白(分别是Smt3和Pmt3)。在哺乳动物中,泛素化涉及2种E1,超过35种E2和超过600种E3酶,而苏木化则由一种异二聚体E1,一种E2(UBC9/UBE2l)和大约10种E3介导。
泛素和苏木通常通过它们的碳端与靶蛋白赖氨酸残基上的εNH2基团发生异肽键连接而与底物结合的。某些情况下,一个靶蛋白结合一个泛素或苏木,另有一些情况下,靶蛋白的不同赖氨酸残基可以分别结合各自的泛素或苏木。此外,由于泛素和某些苏木也具有可修饰的赖氨酸残基,因此反复共轭结合后可形成多聚体链(Berginkand Jentsch, 2009)。对于泛素,氮端甲硫氨酸后的七个赖氨酸残基(Lys6, Lys11, Lys27,Lys29, Lys33, Lys48, 和Lys63)可以被用来形成泛素链。SUMO2/3内部存在可苏木化的赖氨酸残基,因此可形成苏木链。而SUMO1内部不具此等赖氨酸残基,因此可以作为链终结子,被整合到底物上去。这些泛素链与苏木链不仅存在结构与物理特征的差异,也存在功能上的差异。例如,Lys48-,Lys49-,和Lys11-连接的泛素链借到靶蛋白的蛋白酶体降解。而由Lys-63形成的泛素链则与蛋白间互作相关。此外,还有资料表明存在不同赖氨酸残基连接的泛素链 (Komander and Rape, 2012) 与泛素苏木共同连接成的链 (Praefcke et al., 2012) 。
与其它翻译后修饰类似,泛素化与苏木化也是可逆的。虽然苏木靶蛋白异肽键只能被一个小的肽酶家族(SENP1-SENP3和SENP5-SENP7)断裂,但去泛素化酶(DUB)却已经被发现了大约100种。DUB又被进一步分为5种:泛素碳端水解酶(UCH),泛素特异蛋白酶(USP)和卵巢瘤蛋白酶(OTU),约瑟夫素(Josephin),以及Jab1/MPN/Mov34家族(JAMM/MPN+)。其中前四者为半胱氨酸蛋白酶,最后一个是锌离子依赖的金属蛋白酶 (Nijman et al., 2005) 。除了反作用于泛素苏木连接酶活性,DUB和SENP也参与泛素与苏木前体的加工,而且某些DUB是蛋白酶体的内部组分。
�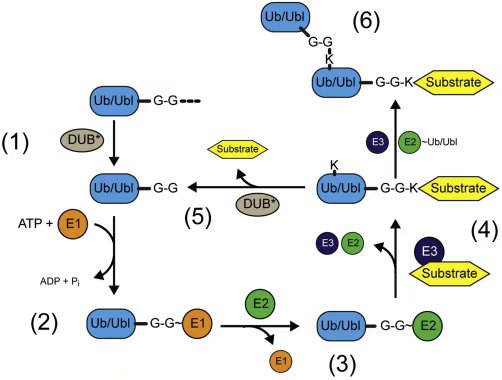 �
�
图一:泛素与类泛素共价结合循环
泛素和苏木最先被翻译为前体多肽,前体多肽首先在碳末端的双甘基序处被加工(1)。之后E1酶利用ATP,将这个基序转换为具有腺苷酸化衍生物的高能键,这个键短暂存在后,立即与E1酶上的一个半胱氨酸结合形成E1-泛素或E1-苏木硫酯中间产物(2)。泛素或苏木反应基团随后通过转酯反应,被转移到E2酶的一个半胱氨酸上,形成E2-泛素或E2-苏木中间产物(3)。多数情况下,E3连接酶作为底物接应子,接应具电荷的E2酶与底物的联系(4)。泛素化和苏木化可以被DUB(对应泛素)或SENP(对应苏木)介导逆向反应(5),否则连续的修饰会形成不同拓扑结构的链(6)。(*)标注其它酶可以移走类泛素(UBL)修饰。
DNA修复与DNA损伤响应
基因组的完整性不断地受到外源性和内源性的DNA损伤化学物、电离辐射(IR)和紫外线(UV)辐射、以及DNA复制错误的破坏。为纾解此困,细胞具有高效的DNA损伤识别、传信和修复的机制,总称为DNA损伤响应(DDR)机制。这些响应机制对细胞和机体生理具有深刻影响,它的损坏和缺失导致的基因组不稳定,与癌症、干细胞耗损、发育缺陷、不育、免疫缺陷、神经退行性疾病以及早衰皆有关联(Jack- son and Bartek, 2009) 。
各种不同的DNA损伤由各自对应的修复机制修复。DNA双链损伤(DSB)被非同源末端连接(NHEJ)、备选NHEJ或者同源重组(HR)三种方式修复。UV导致的DNA损伤,以及其它大块DNA加合物可被核酸切除修复(NER)机制修复。简单的碱基损伤可以被碱基切除修复(BER)机制修复,BER的组分和反应与DNA单链断裂损伤修复重叠。DNA碱基错配可以被错配修复(MMR)机制矫正。DNA的交联,可以被范可尼贫血(FA)途径修复。DNA损伤首先被识别蛋白识别,识别蛋白可以介导并协调后续DNA修复蛋白的募集和活化。DNA损伤会诱发磷酸化、泛素化和苏木化等蛋白翻译后修饰的级联反应,这些反应除了协调前述诸多通路,还会协调DDR的其它方面,包括调节脱氧核糖核酸的供应,引发细胞周期的延迟(细胞周期检验点)。这些事件大多起始于最顶端的DDR激酶ATM和ATR,它们在DDR中的重要性可以从它们的突变疾病,共济失调-毛细血管扩张症(A-T)和赛科尔(Seckel)综合征的表型展现出来 (Durocher andJack- son, 2001; Kerzendorfer and O’Driscoll, 2009)。在这篇综述里,我们关注泛素与苏木在DDR的各个方面被发现的新功能,并着重关注它们响应DSB的机制,因为DSB是所有DNA损伤中细胞毒性最大的。
泛素化与DNA复制后修复
最初将泛素化与DNA修复联系起来,是因为在酵母中发现具有复制后修复(PRR)功能的Rad6蛋白是一种E2泛素酶(Jentschet al., 1987)。PRR是一种DNA损伤容忍性通路,它通过泛素化和苏木化DNA聚合酶延续性因子PCNA来调控DNA复制机器通过大块DNA损伤(图二)。真核生物的PRR具有两个亚途径:跨损伤合成(TLS)和与HR相关的模板切换机制(Ulrich, 2011)。在酵母中,PRR的早期步骤是由E2酶Rad6和指环型(RING-type)E3酶Rad18介导的PCNA的单泛素化 (Hoege et al., 2002; Stelter and Ulrich, 2003)。PCNA最初在Lys164处单泛素化,这一修饰可以被特殊的TLS聚合酶如Polη(伊塔),Polι(妖塔),Polκ(卡帕),和Polζ(泽塔) 识别,这种识别是通过这些聚合酶上的UBM和UBZ家族的泛素结合结构域(UBD),以及与PCNA其它位点互作的基序(如PIP盒)来实现的 (Lehmann, 2011)。与典型的高保真聚合酶不同,TLS聚合酶的催化位点虽然能通过DNA损伤位点,但它却是以序列保真度为代价,使DNA复制产生复制错误倾向。酵母PCNA还可以被E3酶Rad5,联合二聚体型E2酶Ubc13-Mms2,在其Lys164处多聚泛素化。Rad5-Ubc3-Mms2催化形成的PCNA的泛素Lys63链,可以推动与新合成的姐妹染色单体和HR机制相关的模板切换途径修复DNA损伤。虽然目前尚不清楚PCNA泛素化如何推动模板切换,但哺乳动物的ZRANB3转置酶被确定影响PCNA的多泛素化 (Zeman and Cimprich, 2012)。
虽然PRR在酵母与人类之间可能存在差异,不过这一通路大体上是进化上保守的,哺乳动物具有Rad6(人HR6A/UBE2A和HR6B/UBE2B),Rad18(RAD18)和Rad5(SHPRH和HLTF)的同源基因。对于大多数物种,敲除或降低RAD18会导致PRR缺陷、PCNA单泛素化以及在复制叉阻滞点TLS聚合酶如 Polη(伊塔)的积累(Lee and Myung, 2008)。此外RNA干扰研究表明人的HLTF和SHPRH负责PCNA的多泛素化,以响应复制叉诱发的损伤(Motegi et al., 2008) 并抑制突变的发生 (Lin et al., 2011),此表型与E3连接酶在[酵母中]无错PRR中发挥的作用一致。
除了被泛素化,出芽酵母PCNA的Lys164(Lys127程度略低)还可以被E3酶Siz1和E2酶Ubc9苏木化 (Pfander et al., 2005;Stelter and Ulrich, 2003)。PCNA的苏木化,通过募集Srs2可以防止DNA复制过程中盲目的重组。Srs2是一种UvrD型解旋酶,它可以将同源重组关键酶Rad51从染色质上移除 (Krejci et al., 2003; Pfander et al., 2005; Veauteet al., 2003)。Srs2具有PCNA结合结构域PIP和苏木互作基序(SIM),两者都促使Srs2结合苏木化的PCNA (Arm- strong et al.,2012)。虽然人细胞中很难检测到PCNA的苏木化,但是 最近被报道揭示人细胞中存在一个类Srs2蛋白PARI(Moldovan et al., 2012)。
�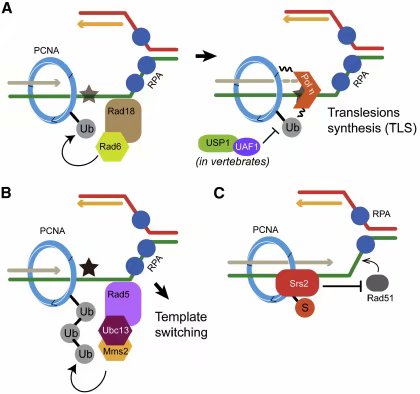 �
�
图二:泛素与苏木在复制后修饰中发挥的作用。(A)PCNA的Lys164处的单泛素化,是在复制叉遇到DNA损伤(图中五角星标记)时获得的。单链DNA被E3连接酶Rad18识别,Rad18与E2酶Rad6一起将PCNA泛素化。泛素化的PCNA募集Y家族DNA聚合酶如 Polη(伊塔),通过跨损伤合成(TLS)过程使复制进程通过损伤位点。在脊椎动物中,去泛素化酶USP1-UAF1可以抗拒PCNA单泛素化。(B)第二种DNA损伤容忍型修复方式是模板切换机制。这种机制在PCNA被E3酶Rad5和二聚体E2酶Ubc13/Mms2多泛素化后发生。模板切换机制需要同源重组。(C)在酵母中,PCNA的Lys164苏木化可以募集Srs2,Srs2可以通过解除Rad51核酸蛋白丝而抑制同源重组的发生。
泛素化调控核酸切除修复(NER)
NER修复大块DNA碱基加合物以及紫外线造成的DNA损伤。先天性NER因子缺陷会导致皮肤色素沉着病(Xeroderma pigmentosum,XP)、磕磕尼综合征(Cockayne syndrome,CS)和毛细血管营养不良(Trichothiodystrophy,TTD)等疾病。这些疾病通常具有日光超敏,皮肤癌倾向(如果为XP的话),认知障碍(cognitiveimpairment),早衰,或者发育不良等特征 (Hoeijmakers, 2009) 。NER包含两大类:泛基因组修复(global genome repair,GG-NER),作用于全部细胞核DNA;伴转录修复(transcription-coupledrepair,TC-NER),特异的作用于转录基因的模板链。在人的GG-NER修复过程中,DNA损伤可以被DDB1-DDB2/XPE和XPC-RAD23这两组复合物各自独立的识别 (Scrima etal., 2011) (图三)。但是之所以认为DDB1-DDB2在NER中起到独特作用,是因为研究者观察到DDB1-DDB2是XPC被募集到染色质上这个过程所需的 (Fitch et al., 2003) 。从机理上讲,DDB1-DDB2以E3链激酶的形式,联合CUL4A/B,介导组蛋白的单泛素化和DDB2与XPC的多泛素化 (Scrima et al., 2011) 。虽然DDB2的自我泛素化使其被靶向降解,而XPC的泛素化则因为受到蛋白酶体互作蛋白RAD23的保护而免于被蛋白酶体降解 (El-Mahdy et al., 2006;Sugasawa, 2006; 以及其中的文献) 。
RNA聚合酶(RNAPII)在遇到DNA损伤导致的转录停滞,可以触发两种独立的级联反应。第一是TC-NER,第二就是将RNAP II泛素化,并将之从染色质上卸下,降解(图三)。TC-NER依赖于与RNAP II关联的SWI/SNF家族的染色质重塑蛋白CSB(ERCC6)(Gaillardand Aguilera, 2013)。除了具有染色质重塑活性,CSB还可将CSA(ERCC8)募集到DNA损伤位点。CSA与DDB1和CUL4一起形成E3连接酶。CSB和CSA的作用可能是对TC-NER进程的许可指令,使得RNAP II后撤与NER核心组分募集等事件得以实现(Gaillard and Aguilera, 2013)。虽然到底哪个泛素化事件启动了TC-NER尚未确认,但RNAP II与CSB的泛素化最有嫌疑。考虑到此,CSB具有一个功能重要的UBD(泛素结合结构域),说明CSB可以识别这一关键泛素化事件 (Anin- dya et al., 2010)。进一步的,DUB(去泛素化酶)USP7被UVSSA(在类CS的UV敏感综合症患者中发生突变)募集到停滞的聚合酶处(Cleaver,2012)。UVSSA-USP7与RNAP II互作,并延迟了CSA依赖的蛋白酶体对CSB的降解。
蛋白酶体降解RNAP II可以被视为TC-NER修复的最终方案,它可以作为一个案例,来研究泛素链的编辑。在酵母中,这些过程涉及E3酶Rsp5(哺乳动物的NEDD4),Rsp5催化RNAPII上泛素的Lys63连接链的形成(Anindya etal., 2007; Wilson et al., 2013)。这种泛素链又可以被一种DUB酶Ubp2剪短,导致RNAP II的单泛素化。而后,单泛素化的RNAPII又可以在Elongin/Culin 3复合体的作用下长出泛素的Lys48连接链,这种链可以在RNAP II被Cdc48分离酶(酵母的VCP/p97同源蛋白)从染色质上卸载下来后,促进RNAPII的降解(Harreman et al., 2009; Verma et al., 2011; Wilsonet al., 2013)。
�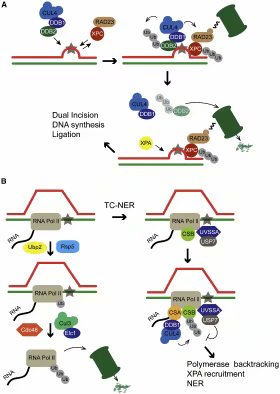 �
�
图三:泛素在核酸切除修复过程中发挥的作用。(A)GG-NER促进基因组DNA上的大块损伤的修复。这种损伤可以被DDB1-DDB2和XPC-RAD23复合体识别。DDB1-DDB2与CUL4和RBX1(未标出)一起组成E3酶,导致DDB2的自我泛素化及其降解,也导致了XPC的多泛素化。XPC可以被RAD23保卫住。RAD23具有类泛素结构域,可以与蛋白酶体互作,使得XPC免于被降解。也正因此,后续的NER过程,如XPA的募集,才得以继续进行。(B)TC-NER修复在转录的DNA链上的大块损伤。当RNA聚合酶在遇到损伤而停滞时,两种独立的修复途径可以发生:第一(右侧),在CSB和CSA发挥了关键作用后,TC-NER被激活。CSB与RNA聚合酶相连,募集CSA,CSA与DDB1组成基于CUL4的E3酶。具体的CSA关联E3酶的关键底物是什么还不清楚,但CSB是被泛素化的,且泛素化的CSB,被由UVSSA募集到停滞的聚合酶处的USP7护卫住,使其降解得以延迟。第二(下部),作为最终方案,RNA聚合酶可以被泛素化,从染色质上卸载,并被蛋白酶体降解。在酵母中,这一步骤由Rad26-Def1(未标出)起始,Rad26-Def1导致Rsp5依赖的RNA聚合酶的泛素化。Rsp5可以促使泛素形成Lys63链,而DUB酶Ubp2可将之剪短为单泛素化。单泛素化的RNA聚合酶成为Elongin-Cullin 3复合物的底物。在RNA聚合酶上新形成的泛素Lys48链,可以协助Cdc48将停滞的聚合酶从染色质上卸下来,RNA聚合酶被卸下来之后被降解。
由RNF8和RNF168组织的基于泛素的DSB传信
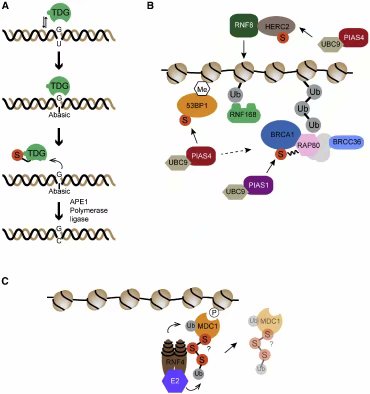
泛素和苏木在细胞内传信的重要例证,是其在募集53BP1和肿瘤抑制蛋白BRCA1到DSB位点外围染色质的过程中展现的协调作用(Lukas et al.,2011)(图四)。这些事件由组蛋白变体H2AX的磷酸化(形成γH2AX)起始的,γH2AX随后被MDC1识别(Stuckiet al., 2005)。MDC1随即被DSB响应激酶ATM磷酸化,这些磷酸化位点与指环型E3连接酶RNF8的FHA结构域结合(Huenet al., 2007; Kolas et al., 2007; Mailand et al., 2007)。此后,RNF8借助与大HECT型连接酶HERC2的互作,将DSB位点的蛋白泛素化(Bekker- Jensen et al., 2010)。随后,指环型E3酶RNF168通过它的UBD结构域(可以识别被RNF8泛素化的底物,以及被其自身泛素化的底物),被募集而来。(The UBDs of RNF168 are notequivalent and are integrated in functional modules containing targetingmotifs, the LRMs that are also present on RAD18 and RAP80)RNF168上的UBD结构域与其它UBD并不相同,它们被整合到包含靶向基序的功能模块(LRM)中,这种LRM也存在于RAD18和RAP80中(Panier et al., 2012)。应对DNA损伤时,RNF8/RNF168依赖的泛素化的结果是将DNA损伤修复或传信蛋白如53BP1、RAD18、BRCA1、RAP80复合体(或称之为BRCA1-A)、HERC2、BMI1、RIF1、RNF169、NPM1、FAAP20以及NIPBL等募集抑或维持在DNA损伤位点周围的染色质上(Lukaset al., 2011)。在正常细胞生理层面,RNF8/RNF168依赖的泛素化,促进了在免疫球蛋白种类切换和非功能性端粒融合过程中的NHEJ(Krackerand Durandy, 2011; Peuscher and Jacobs, 2011; Rai et al., 2011)。除了NHEJ,RNF8也可促进HR。RNF168灭活性突变会在临床上表现出相应DNA损伤修复的缺陷。这种突变发生于与A-T相关的免疫缺陷和细胞辐射敏感综合征RIDDLE的患者上(Stewart et al., 2009)。
�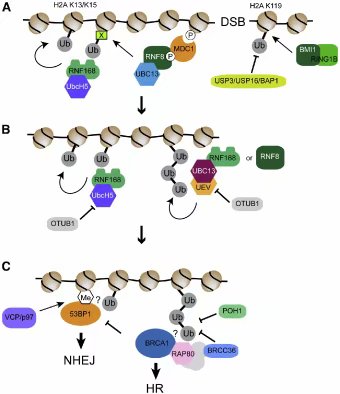 �
�
图四:泛素在基于染色质的DNA双链损伤响应中的作用。(A)DSB导致H2AX的ATM依赖的磷酸化,磷酸化的H2AX被MDC1识别。ATM也磷酸化MDC1,以促进E3连接酶RNF8的募集。RNF8将染色质(X)上的未知因子泛素化,此泛素化随后被RNF168的氮端泛素结合结构域结合。RNF168泛素化H2A,这位RNF168提供了第二个聚集信号。除了RNF8/168,DSB还刺激BMI1-RING1B的募集,这一复合体泛素化H2A的碳端。图中也标出去泛素酶(DUB)对此泛素化过程的抵抗。(B)OTUB1通过与E2酶结合抵抗RNF168依赖的泛素化。RNF8/168也刺激了染色质上63位赖氨酸泛素链的形成。(C)染色质上RNF168依赖的泛素化募集了许多效应因子,包括53BP1(促进NHEJ)和BRCA1(促进HR)。
在有丝分裂时,RNF8途径将会被关闭,这可能与有丝分裂过程中染色质难以泛素化相关(Giuntaet al., 2010)。 In another striking example of regulation, itrecently emerged that RNF168 is a limiting factor in the RNF8 pathway, with theE3 ubiquitin ligases TRIP12 and UBR5 collaborating to regulate RNF168 levels,thereby preventing excessive histone ubiquitylation at DSB sites (Gudjonsson etal., 2012).在另一个引人注目的案例里,泛素连接酶TRIP12与UBR5一起调节RNF168的蛋白含量,从而使RNF168表现为RNF8通路的限速因子,这样可以防止DSB位点过多的组蛋白泛素化(Gudjonsson et al., 2012)。如能揭示TRIP12与UBR5是如何影响RNF168的蛋白含量的,并弄明白有哪些生理条件影响这一调节机制,那这将是非常有意义的一件事。在这点上,TRIP12具有一个WWE结构域,这一结构域在其它蛋白上表现为聚ADP核糖结合活性,因此RNF168的蛋白含量可能受到聚ADP核糖基化的调控。单纯孢疹病毒(Herpes simplex virus,HSV)感染也能调控RNF8通路。HSV的ICP0蛋白(在病毒由潜伏期向裂解期转变的过程中发挥关键作用),是一种E3酶,它可以靶向RNF8和RNF168,导致其降解(Chaurushiya et al.,2012; Lilley et al., 2010)。比较特别的是,ICP0以一种“方向降解子(degron)”的机制,与RNF8的FHA结构域通过磷酸化依赖的方式互作,从而达到靶向降解RNF8的目的(Chaurushiya et al., 2012)。RNF8和RNF168的降解不仅干扰了HSV单链DNA起始的DDR过程,也使得RNF8/RNF168依赖的病毒基因组转录抑制得以解除。
虽然早期工作认为RNF8/168的初级靶蛋白是H2A型组蛋白,不过突变实验发现RNF8/168的靶位点有异于经典的H2A的K119泛素化位点(Huen et al., 2007)。确实,最近的证据显示RNF168靶向H2A的氮端的Lys13/15位点(Gatti et al., 2012;Mattiroli et al., 2012)。一个重要的问题是,我们需证明H2A的Lys13/15的泛素化是否或如何促进DSB位点侧翼染色质对蛋白的募集,以及这种调节怎样与其它的响应DSB的组蛋白修饰相互协调的。对于这点,我们联想到,虽然H2A的Lys13与Lys15,在核小体的立体结构里远离H2A的K119位点,可是它们却与H2B的K120位点离得很近。H2B的K120位点被E3连接酶RNF20和RNF40,在E2连接酶RAD6的协助下泛素化(Zhu et al., 2005)。RNF20与RNF40也是通过ATM依赖的机制,被募集到DSB位点,将H2B单泛素化的(Moyal et al., 2011; Nakamura et al., 2011)。这一联想,可能可以解释为什么Rnf8突变的小鼠胚胎成纤维细胞表现为H2B的K120泛素化正常水平的下降 (Wu et al., 2009)。因为哺乳动物的H2B泛素化可调节转录和染色质凝聚(Weake and Workman, 2008),H2A的K13/K15与H2B的K120的泛素化之间可能的联系,可能在某种程度上解释为何DSB位点附近会发生RNF8/RNF168依赖的转录失活(Shanbhag et al., 2010)。
DNA损伤导致的组蛋白泛素化并不局限在H2A的K13/K15和H2B的K120上。DSB位点也会聚集E3酶BMI1-RING1B,这种E3酶被认为可以提高H2A/H2AX的K119位单泛素化,此种泛素化可能参与DNA损伤诱导的转录沉默 (Gieni et al., 2011; Shanbhag et al., 2010)。此外,蛋白质组学数据揭示,所有核心组蛋白,以及H1、H2AZ、H2AX和macro-H2A在很多位点都能发生泛素化 (Kim et al., 2011; Wagneret al., 2011)。如能发现这些位点的泛素化可以影响DNA损伤或被DNA损伤影响,那这也是很有意思的实验。
53BP1和BRCA1对DSB响应的调控
53BP1和BRCA1是RNF8通路的两个主要效应因子,但它们却具有截然相反的作用:53BP1抑制DNA末端的切割,以此促进DNA损伤的NHEJ修复 (Noon and Goodarzi, 2011),而BRCA1促进DSB的HR修复,并在一定程度上与DNA断裂末端的切割的起始相关 (Li and Greenberg, 2012)。因此53BP1与BRCA1,在NHEJ和HR两种修复机制的拔河比赛中,发挥了关键性的作用。因为53BP1的失活抑制了与BRCA1功能缺失相关的致死、癌变和大多数突变剂的敏感性,此种功能拮抗对我们理解为何BRCA1具有肿瘤抑制因子的功能有很大帮助(Bouwmanet al., 2010; Bunting et al., 2010)。另一种拮抗效果表现在,BRCA1可能是一种53BP1的竞争者或抑制剂,特别是在HR修复明显上调的S/G2期 (Chapman et al., 2012)。
53BP1聚集到DSB位点的机制,到目前还不是很清楚,因为53BP1没有可识别的泛素结合区域。反之,53BP1有一个可以与组蛋白H4的Lys20单或二甲基化(H4K20me1/2)结合的串联都铎结构域(tandem Tudor domain),以及一个与蛋白蛋白互作相关的BRCT结构域 (Botuyanet al., 2006)。需要注意的是,53BP1需要都铎结构域,而不是BRCT区,来完成在DSB位点的聚集 (Pryde et al., 2005)。近来的实验说明,L3MBTL1与JMJD2A两个蛋白平时与H4K20me1/2结合,但是当DNA发生损伤时,这两个蛋白会从染色质上被卸载下来 (Acs et al., 2011; Mallette et al., 2012)。L3MBTL1的卸载需要分离蛋白VCP/p97,VCP/p97可以通过K48连接的泛素与其辅助因子NPL4来靶向聚集到DSB位点 (Acs et al., 2011);而JMJD2则受RNF8通路影响而降解(Mallette et al., 2012)。 Meerang et al. (2011)发现了另外一种机制,也证明VCP/p97参与维持53BP1在DSB位点的定位。虽说单单通过泛素化,就可以将JMJD2A/L3MBTL1卸载的说法很吸引人,但是尚难将这一理论与53BP依赖其结构、其都铎结构域与染色质产生联系的理论(Bothmeret al., 2011),以及H2A的K13/15泛素化在53BP1向DSB位点聚集具有紧要作用的理论(Mattiroliet al., 2012)整合。如果不能在生化层面重建RNF168依赖的53BP1染色质结合,那么就解释不清53BP1向DSB位点聚集的具体机制。
BRCA1与RARD1一起组成一个二聚体的环型E3酶,BRCA1除了作用于HR,也与伴转录DNA修复、DNA交联修复和检验点控制相关。虽然确知BRCA1聚集到DSB位点是凭借RNF8和RNF168依赖的泛素偶合物的形成而完成的 (Lukas et al., 2011),但到底BRCA1是如何识别这一泛素偶合物的还尚在研究当中。BRCA1在DSB位点的长期维持依赖它与RAP80复合体(以及RAP80的泛素结合UIM模块)的互作,但是RAP80对BRCA1向DSB的起始募集是非必需的 (Hu et al., 2011; Yin etal., 2012b)。目前,在BRCA1的E3连接酶活性是否对DNA修复和肿瘤抑制至关重要这个问题的看法上,尚存大量歧见,折中的观点认为这一活性对HR修复并不重要 (Liand Greenberg, 2012; Shakya et al., 2011; Zhu et al., 2011)。确实,当BRCA1的环结构域被突变(BrcaI26A)后,它与E2连接酶的联系被阻断,但是细胞依然可以用HR修复DSB,而且突变鼠表型正常,肿瘤潜伏时间正常(Li and Greenberg, 2012; Shakya et al., 2011)。但是,鸡的DT40细胞的类似突变,导致细胞对拓扑异构酶一毒物喜树碱(camptothecin)引发的DNA损伤极为敏感(Sato et al., 2012)。因此,尚需更多的工作来破解BRCA1的E3连接酶活性的特殊功能。
范可尼贫血通路中的泛素控制
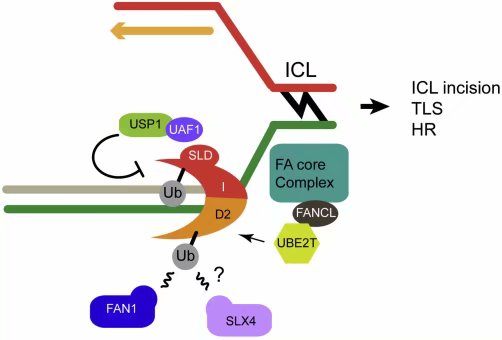
范可尼贫血(FA)是一种罕见的,以发育异常、癌症倾向、慢性骨髓缺陷、以及阻断转录和复制的DNA链间交联(ICL)损伤修复的缺陷等为特征的隐性遗传病 (Garner and Smogorzewska,2011; Kim and D’Andrea, 2012)。目前已知有15个基因(FANCA到FANCP)的双等位基因突变会导致FA,它们对应的蛋白参与S期发生的FA通路的三个基本事件:ICL识别与切除,跨损伤合成与HR介导的修复 (Knipscheeret al., 2009)。复制叉与ICL的相遇,导致FA核心复合体与染色质的结合,这就激活了FA通路(图五)。核心复合体中的FANCL亚基是一个E3连接酶,它与E2酶UBE2T合作,将异二聚体FANCD2-FANCI的两个亚基全部单泛素化,单泛素化的FANCD2-FANCI为后续的修复提供了一个平台(Joo et al., 2011)。特别的,泛素化除了帮助将FANCD2-FANCI挂到染色质上,泛素化的FANCD2也可以被结构特异核酸酶FAN1的名为UBZ4的泛素结合结构域(UBD)识别,也有人说泛素化的FANCD2也可以与核酸酶骨架SLX4(FANCP)互作(Sengerová et al., 2011)。这些因子被募集到DNA损伤位点后,核酸酶剪断损伤的DNA链,引发了后续的包括本综述到处提及的跨损伤合成和HR事件。其它的FA通路与泛素的联系,还包括FA核心复合体相关因子FAAP20借助其自身的UBD与RNF8介导的泛素化的底物结合,与RNF8一起促进FA核心复合体与FANCD2向ICL的募集(Yan et al., 2012)。
在DNA损伤响应中的去泛素酶作用
一个熟知的去泛素酶(DUB)是USP1,它是FANCD2的主要去泛素酶,将其灭活会导致许多范可尼贫血的表征,说明范可尼贫血通路的有效运作既需要控制泛素化,也需要适当的控制去泛素化(Kimand D’Andrea, 2012)。在这点上,USP1的激活因子UAF1/WDR48,具有两个串联的类SUMO结构域,可介导其与FANCI上的SIM相关序列互作,从而将USP1引向FANCD2/FANCI(Yanget al., 2011)(图五)。其它去泛素酶诸如USP3,USP16,BRCC36,POH1和OTUB1都参与RNF8通路的反向调控,其中USP3和USP16就是因为具有反作用于H2A泛素化的活性(Weakeand Workman, 2008),且USP3过表达可阻断RNF168向DSB位点的募集(Doil et al., 2009),才被认为与RNF8通路相关的。在另一方面,USP16反作用于RNF8/168介导的DSB引发的转录抑制(Shanbhaget al., 2010)。BRCC36(BRCC3)是一个JAMM/MPN(+)异肽酶,对63位赖氨酸连接的泛素链具有很强的选择性,它是BRCA1-RAP80复合体的一个组分,可在RNF8/168启动后聚集到DSB位点(Cooper et al., 2009; Donget al., 2003; Shao et al., 2009; Sobhian et al., 2007; Wang and Elledge, 2007)。此外,另一个选择性针对63位赖氨酸泛素链的JAMM/MPN(+)蛋白是蛋白酶体相连的去泛素酶POH1/PSMD14,它与RNF8通路的反向调节相关(Butler et al., 2012)。总结起来,这些发现表明蛋白酶体相连的POH1和BRCA1/RAP80相连的BRCC36可能联合起来限制RNF8/168依赖的63位赖氨酸泛素链在DSB位点形成。值得注意的是,OUT家族去泛素酶OTUB1,虽然是RNF168依赖的泛素化的反向调节子,但奇怪的是,这与它的异肽酶活性是无关的 (Nakada et al., 2010)。相反的,OTUB1是通过它的催化位点与一段伪酶解产物的组合而结合并抑制RNF168相关的E2酶(UBC13/UBE2N和UBE2D/UBE2E家族的E2酶)的(Juang et al., 2012; Wieneret al., 2012)。DUB们无疑还影响了很多其它的DRR过程,比如USP1-UAF1也可调节PCNA的泛素化与TLS(Huang et al., 2006)。其它值得关注的DUB还有:USP47,它可以通过调节DNA聚合酶β的水平而影响BER(Parsonset al., 2011);肿瘤抑制子BAP1,调节H2A的K119ub(Scheuermann et al., 2010);还有USP7,它调节许多DDR蛋白的稳定性,包括p53和ERCC6(Fraile et al., 2012)。由于这些DUB更易于被开发的小分子调控,因此评估这些不同DUB作为药物研发的潜在靶点是比较有意义的事。
� �
�
图五:泛素在范可尼贫血通路中的作用。DNA链间交联(ICL)是可以阻断复制叉的损伤。DNA复制叉的停滞以及FANCM及相关因子(未示)的存在,促进了多亚基的FA核心复合体的募集,这一复合体是一个E3连接酶。这一E3的催化亚基是FANCL(它的E2是UBE2T)。FA核心复合体单泛素化FANCD2-FANCI。FANCD2-FANCI的反诉胡被FAN1(也可能是SLX4)识别,FAN1和SLX4都含有UBZ4型的泛素结合结构域。去泛素酶USP1-UAF1可以反转FANCD2-FANCI的泛素化,这对FA通路甚为重要。核酸酶(FAN1或SLX4)的募集可以切割、解脱ICL,引发后续的TLS或HR。
泛素化调控p53对DNA损伤的响应
脊椎动物的DNA损伤响应的一个主要执行者就是转录因子p53,它的突变与各种类型的人类癌症相关。许多实验室的大量研究都说明p53的含量和功能的调节是为了响应DNA损伤,而泛素化与苏木化在这些调节反应中都起了关键作用。鉴于这方面研究的广度,我们这里只能对它进行粗略的综述(想读全面的综述,请看Laneand Levine, 2010)。简短的说,已发现的参与p53调控的E3酶就超过十个,其中被研究最多的是环型E3酶Mdm2/HDM2(前为小鼠后为人类的基因名)(Brooks and Gu, 2011)。Mdm2可形成同二聚体,也可以与相关E3酶Mdmx/HDMX组成异二聚体,结合并泛素化p53,促进蛋白酶体介导的p53降解,以使p53在正常时保持比较低的含量。然而,当响应DNA损伤时,施加于p53,Mdm2和它们的调节子身上的各类修饰,包括那些被检验点激酶ATM、ATR、CHK1和CHK2催化或促进的,会抑制p53的泛素化和降解,并促进p53的转录活性,使它和它的辅助因子一起促进其靶基因的表达。这些基因又会产生各类响应,包括细胞周期延迟、襄助DNA修复蛋白、以及在某些案例中维持细胞周期停滞或引起凋亡。还有就是,最近的研究强调了各类去泛素酶,包括USP7/HAUSP、USP10、USP29和USP42在调节p53含量和活性上起到了重要作用(Hock et al.,2011)。
BER中苏木化的事例
苏木化与DNA修复的联系首先在BER的研究中被确认(图六)。BER通路由DNA糖苷酶识别各类碱基损伤起始,这些糖苷酶将损伤的碱基移除,留下了无碱基位点,这些位点随后被处理并修复。胸腺嘧啶DNA糖苷酶(TDG)可以移除由5甲基胞嘧啶或胞嘧啶脱氨基产生的G:T或G:U错配中的胸腺嘧啶或尿嘧啶。生化研究表明,与某些其它DNA糖苷酶类似,TDG会强烈的结合到它产生的物碱基位点,TDG与其产物的结合抑制了其自身活性,这会损害BER的后续步骤。值得注意的是,SUMO1与TDG的碳端结合,并与TDG蛋白其它区域的SIM基序发生互作,导致TDG氮端的构象变化,使之与产物结合的能力下降,从而使TDG的酶活性得以恢复(Baba et al., 2005; Steinacher and Scha ̈r, 2005)。这些研究得出一个分子交接模型:未与SUMO连接的TDG结合并接到碱基水解,而之后的SUMO结合与TDG的苏木化,将TDG从靶位点移走。这促进了无碱基BER中间物向APE1核酸内切酶的交接,促使修复过程后续步骤的进行。
DSB修复中的苏木与泛素的对谈
一个被详述的苏木化与DSB响应的直接关联来自对酿酒酵母HR关键因子Rad52的研究。Rad52的苏木化需要Mre11复合体、Ubc9和与哺乳动物的PIAS蛋白(见下文)相关的E3酶Siz2。Rad52的苏木化除了保护Rad52免于被降解,也使Rad52免于进入核仁以防止核糖体DNA重复序列之间的不正常重组(Torres- Rosell et al., 2007)。苏木化也以其它方式控制酵母的HR。黏连素是扣住姐妹染色单体的多蛋白复合体,黏连素扣住姐妹染色单体可以促进有丝分裂时染色体的平均分配,并促进DNA复制后的姐妹染色单体间的同源重组修复。在酿酒酵母中,姐妹染色单体之间的黏连,除了于S期产生,还会在G2/M期因DSB而被局部的和全局的进一步加强。黏连素亚基Mcd1会因DSB的形成而苏木化,这依赖于黏连素相关Smc5-Smc6复合体的组分苏木E3酶Mms21(Nse2),且促进了DNA损伤引发的黏连(McAleenan et al.,2012)。相关通路在脊椎动物中可能也存在,因为人的Smc5-Smc6向DNA损伤位点的聚集(以促进姐妹染色单体同源重组)至少部分的需要MMS21/NSE2对人的SCC1(Mcd1同源蛋白)的苏木化及其对黏连素反向因子WAPL的抵制作用 (Wu et al., 2012)。对酵母中各类HR,NHEJ,BER,NER,MMR以及检验点组分的DNA损伤诱导的苏木化的探究强化了如下问题,苏木化是如何影响各类DDR响应组分的(Cremona et al., 2012)。确实,(Psakhyeand Jentsch, 2012)最近的酿酒酵母的工作揭示,虽然不同HR因子的苏木化集合起来对DSB修复是重要的,但任何单一苏木化的缺失,却只对DSB修复造成轻微影响。这可能是苏木化作用的一个原则,即“蛋白群体修饰”原则,这不仅在DNA修复通路上如此,其它通路也如此。
在哺乳动物中,将苏木化与DSB响应联系在一起,缘起于研究发现SUMO1、SUMO2/3、UBC9以及PIAS和MMS21苏木E3酶可向DSB或复制阻滞位点聚集(Galanty et al., 2009; Morris et al., 2009) (图六)。此外,PIAS4的灭火严重损害了DSB位点RNF168的集聚和63位赖氨酸泛素的累积,以及53BP1和BRCA1的聚集,而PIAS1的确实只阻止了RAP80和BRCA1的聚集。与此一致,PIAS1或PIAS4确实严重损害了HR和NHEJ对DSB的修复,并导致细胞对DSB诱导试剂的高度敏感性(Galanty etal., 2009; Morris et al., 2009)。虽然RNF8或RNF168的确实并不妨碍PIAS1/4向DNA损伤位点的募集,但它显著的减少了SUMO1和SUMO2/3的聚集。这可能是因为RNF8/168介导了BRCA1和53BP1等因子的聚集,而这些因子将会被苏木化,当然,RNF8/168直接作用于PIAS1/4的可能性也是有的。进一步的,虽然PIAS1/4不是RNF8向DAN损伤位点聚集所需要的,但PIAS4(不关PIAS1)的缺失会损害RNF168的聚集。值得注意的是,RNF168和HERC2是以DNA损伤和PIAS4依赖的方式被苏木化的,PIAS4的缺失不仅降低了RNF148的量,也损害了HERC2向RNF8结合,并波坏了IR引发的HERC2向染色质的聚集(Danielsen et al., 2012)。当BRCA1定位到DSB位点后即被PIAS1亦或PIAS4苏木化,这加强了BRCA1的泛素连接酶活性(Morris et al.,2009),这种作用可能是因为苏木化可帮助BRCA1与E2酶亦或其它含有SIM的靶蛋白的关联。PIAS1和PIAS4的其它功能可能是通过其对53BP1等因子的苏木化实现的(Galanty et al., 2009)。苏木E3连接酶在DDR中的功能还表现在如下两点:PIAS1余SNM1A之间的互作促进了ICL的修复 (Ishiai et al., 2004),酪氨酸DNA磷酸二酯酶(TDP1)的苏木化增强DNA单链断裂的修复 (Hudson et al., 2012)。此外,虽然在S期SUMO蛋白酶SENP6可与RPA70互作,保持RPA70低苏木化,但DNA复制的压迫将这一复合体解离,允许SUMO2/3修饰的RPA70的积累,苏木化RPA70的积累又可以将RAD51募集到损伤的DNA处,从而促进HR (Dou et al., 2010) 。在人的布鲁姆综合征(Bloom’ssyndrome)存在缺陷的RecQ家族DNA解旋酶BLM,也可以被苏木化,如引入突变,阻断BLM之苏木化,就会引起S期DNA损伤的产生、DNA损伤的高度敏感性并损害RAD51向复制阻滞位点的定位 (Ouyang et al., 2009) 。
� �
�
图六:苏木在各类DNA损伤响应通路中的作用之实例。(1)BER中的胸腺嘧啶DNA糖苷酶(TDG)被它的产物抑制。TDG的苏木化使其构象发生变化,从而使其从其产物上解脱下来。(B)在基于染色质的DSB响应中的,苏木化在各步骤里的作用。详见正文。(C)苏木化也可以促进泛素化。比如STUbL蛋白,它是一类E3泛素连接酶,可通过识别被苏木化的底物上的苏木将底物泛素化。在实例中,RNF4在DSB响应时促进MDC1的周转(或重复利用)。
其它泛素化与苏木化之间的联系可以通过靶向苏木的泛素连接酶(STUbL)来说明,它们包括人的RNF4,裂殖酵母的Rfp1/2-Slx8和酿酒酵母的Slx5-Slx8(Prudden et al., 2007) 。STUbL含有SIM基序,可以结合靶蛋白上的耦合的苏木或者其中的类苏木结构域,然后将靶蛋白泛素化,这通常会导致靶蛋白被蛋白酶降解。虽然研究表明酿酒酵母的Slx5-Slx8和裂殖酵母的Rfp1/2-Slx8调节HR,但是其具体机制至今未明。相反,近期研究显示,在人和鸡的细胞中,RNF4的灭活不仅引起DSB的HR修复缺陷,也导致NHEJ修复的缺陷(Galanty et al., 2012; Luo et al., 2012; Yin et al., 2012a) 。还有就是,RNF4向DSB位点的募集是依赖它自身氮端的SIM基序以及被苏木化的DSB响应蛋白,诸如53BP1,MDC1和RPA,被募集之后,RNF4介导靶蛋白的泛素化的积累(图六)。虽然RNF4的功能底物有很多,但其中主要的应该是MDC1和RPA,RNF4可以促进这些底物在DSB位点的周转率。例如,由RNF4缺失或RPA苏木化位点的突变造成的RPA周转率下降,会导致RAD51替换RPA的效率下降,因而损害HR(Galanty et al.,2012)。因为RNF4促进蛋白酶体向DSB位点的聚集,如将蛋白酶体的亚基去除,就会引起与RNF4缺失类似的表型,这说明RNF4介导的靶向苏木的DDR因子的泛素化导致这些底物被蛋白酶体识别,以此造成它们在所在位置的周转,以协助DNA损伤修复各个阶段的逐步运行。考虑到此,裂殖酵母VCP/p97的对应蛋白Cdc48,通过它的辅因子Ufd1的一个SIM基序,与SUMO偶合物结合(Nie et al., 2012)。因为VCP也可以与泛素结合, VCP可能通过与STUbL的靶蛋白(被泛素和苏木双重修饰)的结合促进DNA修复。既然酿酒酵母的Ufd1也可结合苏木且人类的VCP/p97在DSB位点发挥作用(见前文),那么,STUbL靶蛋白、VCP/p97复合体和蛋白酶体之间的联系可能在真核生物细胞中都存在。
运用于治病
致DNA损伤的化学疗法与辐射疗法被广泛用于癌症治疗且大多有效,它们的效果反映了它们对癌细胞造成的DNA损伤的致死强度。不幸的是, 由于DNA损伤对正常组织有损害且涉及毒理反应,这限制了治疗癌症时施加于癌细胞的DNA损伤的程度,因为程度不足,癌细胞内在机制经常会对这些治疗性DNA损伤产生耐性,或者将之修复,致使癌症复发。致DNA损伤药物治疗癌症的疗效,可能是因为癌细胞较大多数正常细胞增殖要快,因此会面对更大的DNA损伤负担,也因为它们某一DDR成分受损而使它们特别的依赖这一组分(Helleday et al., 2008; Jackson and Bartek,2009)。除了可以改善致DNA损伤疗法在特种癌症上疗效,对癌细胞与正常细胞DDR差异的研究,也为以组合致死为理论基础的癌细胞选择性消灭的疗法提供待选的靶点。组合致死概念最初是从PARP1/2抑制剂杀死HR缺陷(BRCA1/2突变)的肿瘤的实验阐述出来的(Bryantet al., 2005; Farmer et al., 2005)。对泛素化和苏木化如何影响DDR过程的理解的加深,可能为我们控制肿瘤提供新方案。例如,遗传导致的泛素化缺陷可致DDR缺陷,从这可以推测癌症演变的过程中泛素化的缺陷可能在体细胞中发生,而这就提供了新的检测靶点和可能的治疗靶点。进一步的,泛素和苏木系统的一系列蛋白,其中许多可作为天然的药物酶,这为新药物靶点的研发提供了大量的未开发资源。值得注意的是,硼替佐米(bortezomib(完克Velcade))可以使细胞对放射治疗和致DNA损伤化学治疗更加敏感 (Motegi et al., 2009) ,而且近期研究揭示完克能造成多种骨髓瘤细胞( myeloma cells)处于HR受损状态,因而使细胞高敏于PARP抑制剂,这点也说明多种试剂组合使用可能具有潜在临床价值(Neri et al., 2011) 。研发靶向泛素苏木系统的DDR特异组分的试剂可能也具有抗癌能力,其中,针对某些DDR事件的靶蛋白的试剂,可能比那些针对泛素蛋白酶体系统的一般性抑制剂的毒性更小。对泛素苏木与DDR的了解,也可能为DNA损伤亦或DDR失调所致衰老相关疾病的诊断和治疗带来更好的方案。
展望
过去的几年见证了我们对泛素和苏木偶合物调控DNA损伤响应的研究结果的急速积累。这一积累因我们对泛素和苏木系统的普遍认知的加深、新技术方法的涌现、以及参与这一重要领域研究的大量科技工作者而加速。除了鉴定参与DNA修复和相关事件的泛素和苏木系统的组分,另一未来研究的重要领域就是评判其它类泛素调节子是否或如何参与到DDR的。而在未来的挑战中,我们认为最重要的工作将会是清晰的阐明泛素化、苏木化和相关事件如何控制DDR进程。如前文所讲,某些时候,泛素和苏木除了通过单体耦合、单独形式或两者组合的方式控制DDR外,也可以通过不同的连接方式组成针对特殊靶蛋白的不同形式的链拓扑结构。已知的证据指出,这些不同的生物学特异作用是通过被泛素苏木修饰的蛋白被解读因子结合实现的,这些解读因子包含泛素/类泛素结合基序和靶向序列,因此它们可以在特异蛋白底物的情况下识别特殊的泛素/苏木链或链间键。泛素/苏木/类泛素密码的生化和功能多样性的程度确实是极大的,而如果将可能与这些修饰一同起作用的磷酸化和聚二磷酸腺苷核糖基化( poly(ADP) ribosylation )整合起来的话,其多样性程度就更大了。我们对泛素化或苏木化的认知中与DDR相关的其它领域还有VCP/p97的功能以及泛素链的编辑。鉴于其深度和广度,DDR研究共同体可能还将发现关于泛素化和苏木化的更多的原则和实例,其中的很多也会出现于被此二物翻译后修饰控制的其它细胞活动中。
致谢
对未曾引用亦或因篇幅限制而未引之重要发现之作者深以为歉。对某,某,某,某,某,某和某之版前交流、建议亦或讨论深以为谢。杰克逊史蒂芬实验室受联合王国癌症研究(CRUK)项目之某号,欧洲研究协会( European Research Council )之某号以及欧洲共同体第七框架计划之某号之资助。核心基础设施资金由CRUK之某号及惠康信托之某号资助。杰克逊史蒂芬由剑桥大学支付薪水,CRUK提供补贴。 堵罗赫丹尼尔实验室对泛素和DNA损伤的研究由CIHR之某号资助。堵罗赫丹尼尔为D关于NA损伤响应的分子遗传学加拿大研究协会(CanadaResearch Chair)主席(一级)也是癌症发生机理研究的季兰思托马斯主席(Thomas KieransChair)。杰克逊史蒂芬无利益纠纷,但他透漏他是使命治疾有限公司(Mission TherapeuticsLtd)的创始人及股东。
参考文献
Huen, M.S., Grant, R.,Manke, I., Minn, K., Yu, X., Yaffe, M.B., and Chen, J. (2007). RNF8 transducesthe DNA-damage signal via histone ubiquitylation and checkpoint proteinassembly. Cell 131, 901–914.
Ishiai, M., Kimura, M.,Namikoshi, K., Yamazoe, M., Yamamoto, K., Arakawa, H., Agematsu, K., Matsushita,N., Takeda, S., Buerstedde, J.M., and Takata, M. (2004). DNA cross-link repairprotein SNM1A interacts with PIAS1 in nuclear focus formation. Mol. Cell. Biol.24, 10733–10741.
Jackson, S.P., and Bartek,J. (2009). The DNA-damage response in human biology and disease. Nature 461,1071–1078.
Jentsch, S., McGrath, J.P.,and Varshavsky, A. (1987). The yeast DNA repair gene RAD6 encodes aubiquitin-conjugating enzyme. Nature 329, 131–134.
Joo, W., Xu, G., Persky,N.S., Smogorzewska, A., Rudge, D.G., Buzovetsky, O., Elledge, S.J., andPavletich, N.P. (2011). Structure of the FANCI-FANCD2 complex: insights intothe Fanconi anemia DNA repair pathway. Science 333, 312–316.
Juang, Y.C., Landry, M.C.,Sanches, M., Vittal, V., Leung, C.C., Ceccarelli, D.F., Mateo, A.R., Pruneda,J.N., Mao, D.Y., Szilard, R.K., et al. (2012). OTUB1 co-opts Lys48-linkedubiquitin recognition to suppress E2 enzyme function. Mol. Cell 45, 384–397.
Kerzendorfer, C., andO’Driscoll, M. (2009). Human DNA damage response and repair deficiency syndromes:linking genomic instability and cell cycle checkpoint proficiency. DNA Repair(Amst.) 8, 1139–1152.
Kim, H., and D’Andrea, A.D.(2012). Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway.Genes Dev. 26, 1393–1408.
Kim, W., Bennett, E.J.,Huttlin, E.L., Guo, A., Li, J., Possemato, A., Sowa, M.E., Rad, R., Rush, J.,Comb, M.J., et al. (2011). Systematic and quantitative assessment of theubiquitin-modified proteome. Mol. Cell 44, 325–340.
Knipscheer, P., Ra ̈ schle,M., Smogorzewska, A., Enoiu, M., Ho, T.V., Scha ̈ rer, O.D., Elledge, S.J., andWalter, J.C. (2009). The Fanconi anemia pathway promotes replication-dependentDNA interstrand cross-link repair. Science 326, 1698–1701.
Kolas, N.K., Chapman, J.R.,Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F.D., Panier, S., Mendez, M.,Wildenhain, J., Thomson, T.M., et al. (2007). Orchestration of the DNA-damageresponse by the RNF8 ubiquitin ligase. Science 318, 1637–1640.
Komander, D., and Rape, M.(2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229.
Kracker, S., and Durandy,A. (2011). Insights into the B cell specific process of immunoglobulin classswitch recombination. Immunol. Lett. 138, 97–103.
Krejci, L., Van Komen, S.,Li, Y., Villemain, J., Reddy, M.S., Klein, H., Ellen- berger, T., and Sung, P.(2003). DNA helicase Srs2 disrupts the Rad51 presyn- aptic filament. Nature423, 305–309.
Lane, D., and Levine, A.(2010). p53 Research: the past thirty years and the next thirty years. ColdSpring Harb. Perspect. Biol. 2, a000893.
Lee, K.Y., and Myung, K.(2008). PCNA modifications for regulation of post- replication repair pathways.Mol. Cells 26, 5–11.
Lehmann, A.R. (2011).Ubiquitin-family modifications in the replication of DNA damage. FEBS Lett.585, 2772–2779.
Li, M.L., and Greenberg,R.A. (2012). Links between genome integrity and BRCA1 tumor suppression. TrendsBiochem. Sci. 37, 418–424.
Lilley, C.E., Chaurushiya,M.S., Boutell, C., Landry, S., Suh, J., Panier, S., Everett, R.D., Stewart,G.S., Durocher, D., and Weitzman, M.D. (2010). A viral E3 ligase targets RNF8and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J.29, 943–955.
Lin, J.R., Zeman, M.K.,Chen, J.Y., Yee, M.C., and Cimprich, K.A. (2011). SHPRH and HLTF act in adamage-specific manner to coordinate different forms of postreplication repairand prevent mutagenesis. Mol. Cell 42, 237–249.
Lukas, J., Lukas, C., andBartek, J. (2011). More than just a focus: The chro- matin response to DNAdamage and its role in genome integrity maintenance. Nat. Cell Biol. 13,1161–1169.
Luo, K., Zhang, H., Wang,L., Yuan, J., and Lou, Z. (2012). Sumoylation of MDC1 is important for properDNA damage response. EMBO J. 31, 3008– 3019.
Mailand, N., Bekker-Jensen,S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., and Lukas, J. (2007).RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assemblyof repair proteins. Cell 131, 887–900.
Mallette, F.A., Mattiroli,F., Cui, G., Young, L.C., Hendzel, M.J., Mer, G., Sixma, T.K., and Richard, S.(2012). RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1recruitment to DNA damage sites. EMBO J. 31, 1865–1878.
Mattiroli, F., Vissers,J.H., van Dijk, W.J., Ikpa, P., Citterio, E., Vermeulen, W., Marteijn, J.A.,and Sixma, T.K. (2012). RNF168 ubiquitinates K13-15 on H2A/ H2AX to drive DNAdamage signaling. Cell 150, 1182–1195.
McAleenan, A.,Cordon-Preciado, V., Clemente-Blanco, A., Liu, I.C., Sen, N., Leonard, J.,Jarmuz, A., and Arago ́ n, L. (2012). SUMOylation of the a-kleisin subunit ofcohesin is required for DNA damage-induced cohesion. Curr. Biol. 22, 1564–1575.
Meerang, M., Ritz, D.,Paliwal, S., Garajova, Z., Bosshard, M., Mailand, N., Janscak, P., Hu ̈ bscher,U., Meyer, H., and Ramadan, K. (2011). The ubiqui- tin-selective segregaseVCP/p97 orchestrates the response to DNA double- strand breaks. Nat. Cell Biol.13, 1376–1382.
Moldovan, G.L., Dejsuphong,D., Petalcorin, M.I., Hofmann, K., Takeda, S., Boulton, S.J., and D’Andrea,A.D. (2012). Inhibition of homologous recombina- tion by the PCNA-interactingprotein PARI. Mol. Cell 45, 75–86.
Morris, J.R., Boutell, C.,Keppler, M., Densham, R., Weekes, D., Alamshah, A., Butler, L., Galanty, Y.,Pangon, L., Kiuchi, T., et al. (2009). The SUMO modifi- cation pathway isinvolved in the BRCA1 response to genotoxic stress. Nature 462, 886–890.
Motegi, A., Liaw, H.J.,Lee, K.Y., Roest, H.P., Maas, A., Wu, X., Moinova, H., Markowitz, S.D., Ding,H., Hoeijmakers, J.H., and Myung, K. (2008). Poly- ubiquitination ofproliferating cell nuclear antigen by HLTF and SHPRH prevents genomicinstability from stalled replication forks. Proc. Natl. Acad. Sci. USA 105,12411–12416.
Motegi, A., Murakawa, Y.,and Takeda, S. (2009). The vital link between the ubiquitin-proteasome pathwayand DNA repair: impact on cancer therapy. Cancer Lett. 283, 1–9.
Moyal, L., Lerenthal, Y.,Gana-Weisz, M., Mass, G., So, S., Wang, S.Y., Eppink, B., Chung, Y.M., Shalev,G., Shema, E., et al. (2011). Requirement of ATM-dependent monoubiquitylationof histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 41,529–542.
Nakada, S., Tai, I.,Panier, S., Al-Hakim, A., Iemura, S., Juang, Y.C., O’Donnell, L., Kumakubo, A.,Munro, M., Sicheri, F., et al. (2010). Non-canonical inhibition of DNAdamage-dependent ubiquitination by OTUB1. Nature 466, 941–946.
Nakamura, K., Kato, A.,Kobayashi, J., Yanagihara, H., Sakamoto, S., Oliveira, D.V., Shimada, M.,Tauchi, H., Suzuki, H., Tashiro, S., et al. (2011). Regulation of homologousrecombination by RNF20-dependent H2B ubiquitination. Mol. Cell 41, 515–528.
Neri, P., Ren, L., Gratton,K., Stebner, E., Johnson, J., Klimowicz, A., Duggan, P., Tassone, P., Mansoor,A., Stewart, D.A., et al. (2011). Bortezomib-induced ‘‘BRCAness’’ sensitizesmultiple myeloma cells to PARP inhibitors. Blood 118, 6368–6379.
Nie, M., Aslanian, A.,Prudden, J., Heideker, J., Vashisht, A.A., Wohlschlegel, J.A., Yates, J.R.,3rd, and Boddy, M.N. (2012). Dual recruitment of Cdc48 (p97)-Ufd1-Npl4ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO)and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stabilityfunctions. J. Biol. Chem. 287, 29610–29619.
Nijman, S.M., Luna-Vargas,M.P., Velds, A., Brummelkamp, T.R., Dirac, A.M., Sixma, T.K., and Bernards, R.(2005). A genomic and functional inventory of deubiquitinating enzymes. Cell123, 773–786.
Noon, A.T., and Goodarzi,A.A. (2011). 53BP1-mediated DNA double strand break repair: insert bad punhere. DNA Repair (Amst.) 10, 1071–1076.
Ouyang, K.J., Woo, L.L.,Zhu, J., Huo, D., Matunis, M.J., and Ellis, N.A. (2009). SUMO modificationregulates BLM and RAD51 interaction at damaged repli- cation forks. PLoS Biol.7, e1000252.
Panier, S., Ichijima, Y.,Fradet-Turcotte, A., Leung, C.C., Kaustov, L., Arrow- smith, C.H., and Durocher,D. (2012). Tandem protein interaction modules organize the ubiquitin-dependentresponse to DNA double-strand breaks. Mol. Cell 47, 383–395.
Parsons, J.L., Dianova,I.I., Khoronenkova, S.V., Edelmann, M.J., Kessler, B.M., and Dianov, G.L.(2011). USP47 is a deubiquitylating enzyme that regu- lates base excisionrepair by controlling steady-state levels of DNA poly- merase b. Mol. Cell 41,609–615.
Peuscher, M.H., and Jacobs,J.J. (2011). DNA-damage response and repair activities at uncapped telomeresdepend on RNF8. Nat. Cell Biol. 13, 1139– 1145.
Pfander, B., Moldovan,G.L., Sacher, M., Hoege, C., and Jentsch, S. (2005). SUMO-modified PCNArecruits Srs2 to prevent recombination during S phase. Nature 436, 428–433.
Praefcke, G.J., Hofmann,K., and Dohmen, R.J. (2012). SUMO playing tag with ubiquitin. Trends Biochem.Sci. 37, 23–31.
Prudden, J., Pebernard, S.,Raffa, G., Slavin, D.A., Perry, J.J., Tainer, J.A., McGowan, C.H., and Boddy,M.N. (2007). SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26,4089–4101.
Pryde, F., Khalili, S.,Robertson, K., Selfridge, J., Ritchie, A.M., Melton, D.W., Jullien, D., andAdachi, Y. (2005). 53BP1 exchanges slowly at the sites of DNA damage andappears to require RNA for its association with chromatin. J. Cell Sci. 118,2043–2055.
Psakhye, I., and Jentsch,S. (2012). Protein group modification and synergy in the SUMO pathway asexemplified in DNA repair. Cell 151, 807–820.
Rai, R., Li, J.M., Zheng,H., Lok, G.T., Deng, Y., Huen, M.S., Chen, J., Jin, J., and Chang, S. (2011).The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere endprotection. Nat. Struct. Mol. Biol. 18, 1400–1407.
Sato, K., Sundaramoorthy,E., Rajendra, E., Hattori, H., Jeyasekharan, A.D., Ayoub, N., Schiess, R.,Aebersold, R., Nishikawa, H., Sedukhina, A.S., et al. (2012). A DNA-damageselective role for BRCA1 E3 ligase in claspin ubiquity- lation, CHK1activation, and DNA repair. Curr. Biol. 22, 1659–1666.
Scheuermann, J.C., de AyalaAlonso, A.G., Oktaba, K., Ly-Hartig, N., McGinty, R.K., Fraterman, S., Wilm,M., Muir, T.W., and Mu ̈ ller, J. (2010). Histone H2A deubiquitinase activityof the Polycomb repressive complex PR-DUB. Nature 465, 243–247.
Scrima, A., Fischer, E.S.,Lingaraju, G.M., Bo ̈ hm, K., Cavadini, S., and Thoma ̈ , N.H. (2011).Detecting UV-lesions in the genome: The modular CRL4 ubiquitin ligase does itbest!. FEBS Lett. 585, 2818–2825.
Sengerova ́, B., Wang,A.T., and McHugh, P.J. (2011). Orchestrating the nucleases involved in DNAinterstrand cross-link (ICL) repair. Cell Cycle 10, 3999–4008.
Shakya, R., Reid, L.J.,Reczek, C.R., Cole, F., Egli, D., Lin, C.S., deRooij, D.G., Hirsch, S., Ravi,K., Hicks, J.B., et al. (2011). BRCA1 tumor suppression depends on BRCTphosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528.
Shanbhag, N.M.,Rafalska-Metcalf, I.U., Balane-Bolivar, C., Janicki, S.M., and Greenberg, R.A.(2010). ATM-dependent chromatin changes silence transcrip- tion in cis to DNAdouble-strand breaks. Cell 141, 970–981.
Shao, G., Lilli, D.R.,Patterson-Fortin, J., Coleman, K.A., Morrissey, D.E., and Greenberg, R.A.(2009). The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizesRNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc.Natl. Acad. Sci. USA 106, 3166–3171.
Sobhian, B., Shao, G.,Lilli, D.R., Culhane, A.C., Moreau, L.A., Xia, B., Living- ston, D.M., andGreenberg, R.A. (2007). RAP80 targets BRCA1 to specific ubiquitin structures atDNA damage sites. Science 316, 1198–1202.
Steinacher, R., and Schär, P. (2005). Functionality of human thymine DNA glycosylase requiresSUMO-regulated changes in protein conformation. Curr. Biol. 15, 616–623.
Stelter, P., and Ulrich,H.D. (2003). Control of spontaneous and damage- induced mutagenesis by SUMO andubiquitin conjugation. Nature 425, 188–191.
Stewart, G.S., Panier, S.,Townsend, K., Al-Hakim, A.K., Kolas, N.K., Miller, E.S., Nakada, S., Ylanko,J., Olivarius, S., Mendez, M., et al. (2009). The RIDDLE syndrome proteinmediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell136, 420–434.
Stucki, M., Clapperton,J.A., Mohammad, D., Yaffe, M.B., Smerdon, S.J., and Jackson, S.P. (2005). MDC1directly binds phosphorylated histone H2AX to regulate cellular responses toDNA double-strand breaks. Cell 123, 1213– 1226.
Sugasawa, K. (2006).UV-induced ubiquitylation of XPC complex, the UV- DDB-ubiquitin ligase complex,and DNA repair. J. Mol. Histol. 37, 189–202.
Torres-Rosell, J.,Sunjevaric, I., De Piccoli, G., Sacher, M., Eckert-Boulet, N., Reid, R.,Jentsch, S., Rothstein, R., Arago ́n, L., and Lisby, M. (2007). The Smc5-Smc6complex and SUMO modification of Rad52 regulates recombina- tional repair atthe ribosomal gene locus. Nat. Cell Biol. 9, 923–931.
Ulrich, H.D. (2011). Timingand spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett. 585,2861–2867.
Veaute, X., Jeusset, J.,Soustelle, C., Kowalczykowski, S.C., Le Cam, E., and Fabre, F. (2003). The Srs2helicase prevents recombination by disrupting Rad51 nucleoprotein filaments.Nature 423, 309–312.
Verma, R., Oania, R., Fang,R., Smith, G.T., and Deshaies, R.J. (2011). Cdc48/ p97 mediates UV-dependentturnover of RNA Pol II. Mol. Cell 41, 82–92.
Wagner, S.A., Beli, P.,Weinert, B.T., Nielsen, M.L., Cox, J., Mann, M., and Choudhary, C. (2011). Aproteome-wide, quantitative survey of in vivo ubiqui- tylation sites revealswidespread regulatory roles. Mol. Cell. Proteomics 10, M111, 013284.
Wang, B., and Elledge, S.J.(2007). Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 104, 20759–20763.
Weake, V.M., and Workman,J.L. (2008). Histone ubiquitination: triggering gene activity. Mol. Cell 29,653–663.
Wiener, R., Zhang, X.,Wang, T., and Wolberger, C. (2012). The mechanism of OTUB1-mediated inhibitionof ubiquitination. Nature 483, 618–622.
Wilson, M.D., Harreman, M.,and Svejstrup, J.Q. (2013). Ubiquitylation and degradation of elongating RNApolymerase II: The last resort. Biochim. Bio- phys. Acta 1829, 151–157.
Wu, J., Huen, M.S., Lu,L.Y., Ye, L., Dou, Y., Ljungman, M., Chen, J., and Yu, X. (2009). Histoneubiquitination associates with BRCA1-dependent DNA damage response. Mol. Cell.Biol. 29, 849–860.
806 Molecular Cell 49,March 7, 2013 a2013 Elsevier Inc.
Wu, N., Kong, X., Ji, Z.,Zeng, W., Potts, P.R., Yokomori, K., and Yu, H. (2012). Scc1 sumoylation byMms21 promotes sister chromatid recombination through counteracting Wapl. GenesDev. 26, 1473–1485.
Yan, Z., Guo, R.,Paramasivam, M., Shen, W., Ling, C., Fox, D., 3rd, Wang, Y., Oostra, A.B.,Kuehl, J., Lee, D.Y., et al. (2012). A ubiquitin-binding protein, FAAP20, linksRNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Mol.Cell 47, 61–75.
Yang, K., Moldovan, G.L.,Vinciguerra, P., Murai, J., Takeda, S., and D’Andrea, A.D. (2011). Regulationof the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 25,1847–1858.
Yin, Y., Seifert, A., Chua,J.S., Maure, J.F., Golebiowski, F., and Hay, R.T. (2012a). SUMO-targetedubiquitin E3 ligase RNF4 is required for the response of human cells to DNAdamage. Genes Dev. 26, 1196–1208.
Yin, Z., Menendez, D.,Resnick, M.A., French, J.E., Janardhan, K.S., and Jetten, A.M. (2012b). RAP80is critical in maintaining genomic stability and suppressing tumor development.Cancer Res. 72, 5080–5090.
Zeman, M.K., and Cimprich,K.A. (2012). Finally, polyubiquitinated PCNA gets recognized. Mol. Cell 47,333–334.
Zhu, B., Zheng, Y., Pham,A.D., Mandal, S.S., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2005).Monoubiquitination of human histone H2B: the factors involved and their rolesin HOX gene regulation. Mol. Cell 20, 601–611.
Zhu, Q., Pao, G.M., Huynh,A.M., Suh, H., Tonnu, N., Nederlof, P.M., Gage, F.H., and Verma, I.M. (2011).BRCA1 tumour suppression occurs via hetero- chromatin-mediated silencing.Nature 477, 179–184.
https://m.sciencenet.cn/blog-331314-1133597.html
上一篇:Bioconductor中DESeq2的用法
下一篇:内部翻译资料:癌生物学中之DNA损伤与细胞生死抉择