博文
内部翻译资料:癌生物学中之DNA损伤与细胞生死抉择
||||
癌生物学中之DNA损伤与细胞生死抉择
《癌症自然综述》
(DNA damage and the balance between survival anddeath in cancer biology)
如斯卫南德,托马斯阿达木和凯纳本德
德国美因茨之大学医疗中心之毒理研究所
通讯至凯纳本德,kaina@uni-mainz.de
摘要:DNA可因内源性代谢物、环境与食物中之致癌物、某些消炎药以及基因毒性癌症疗法的作用而发生损伤。细胞通过激活复合体传信网络来响应DNA损伤,以此决定细胞命运,它不仅能促使DNA修复和细胞存活,也可促使细胞死亡。DNA损伤后细胞对于生死之抉择,依赖于DNA损伤识别修复和耐受相关的因子,也依赖于对凋亡、坏死、自噬和大限( apoptosis, necrosis,autophagy and senescence,译者注:大限,是指正常细胞在体外分裂若干次后,即停止分裂,坐以待亡 )通路的激活。这些决定细胞命运的通路或纠缠于或有大助于癌症的起始和发展。进一步的,它们决定了基因毒性药物治疗癌症的效果。理解这些通路的分子基础不仅有助于加深对癌症发生的理解,也可改善癌症治疗的功效。在本综述里,我们记述了DNA损伤后影响细胞生死抉择的关键决策节点。
环境中之化学致癌物、细胞或肠道微生物代谢产生的体内致癌物[1]、活化的免疫细胞诸如单核细胞和巨噬细胞产生的自由基[2],紫外线(UV)辐射[3],电离辐射[4],以及很多药品,特别是基因毒性抗癌药[6],可攻击DNA,造成各类DNA损伤(图一a)。这些损伤造成的基因突变和染色体毁伤,乃癌变和长瘤之要因[7]。为控制基因组的不稳定性,细胞具有DNA损伤响应机制以及DNA修复蛋白,来移除或忍受DNA损伤[8]。如损伤不能被修复,就会具有毒性,促进诸如凋亡和坏死的细胞消亡通路[9],而凋亡与坏死等通路在肿瘤抑制上也有其功用[10]。DNA损伤后细胞死亡是一个被调控的过程,它是借助于求生因子和自尽因子相较的门限来评判的,对细胞生与死的抉择[11]。理解细胞如何以死抗伤,对开发基因毒性化学疗法杀死肿瘤细胞具有重大价值。
�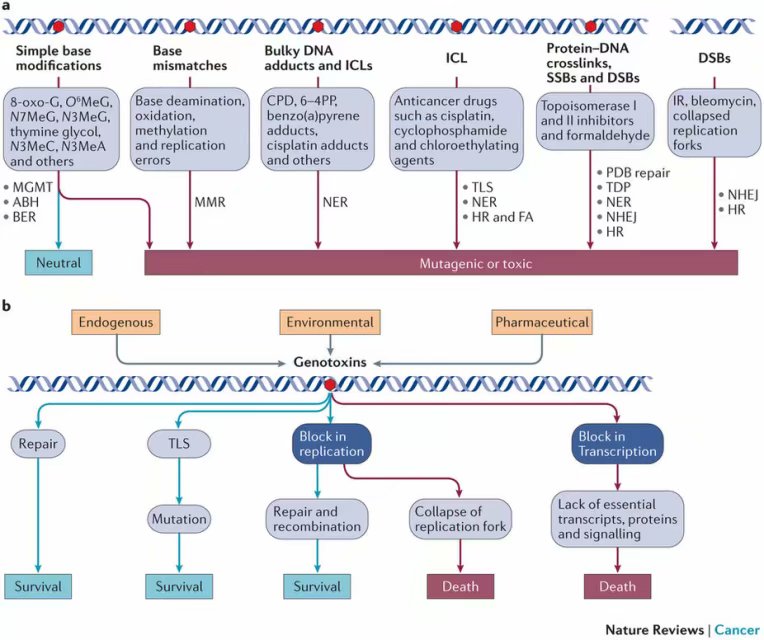 �
�
图一:DNA损伤的细胞对策。(a)简单的碱基修饰被O6MeGDNA甲基转移酶(MGMT)、alkB同源蛋白(ABH)家族成员和碱基外切修复(BER)纠正。碱基错配被错配修复(MMR)修复。大块加合物被核酸外切修复(NER)移除。链间和链内交联(ICL)被跨损伤合成(TLS)、NER、同源重组(HR)和范可尼贫血(FA)通路联合修复。蛋白DNA交联被蛋白连接的DNA断裂(PDB)修复机制、NER、非同源末端连接(NHEJ)和HR联合修复。DNA双链损伤(DSB)被NHEJ和HR修复。未修复的DNA损伤可能是中性的、突变型的或毒性的。(b)致成DNA损伤之因由甚多,或导致DNA之突变或重组,或干扰DNA的复制或转录。根据损伤类型和细胞类型的不同,这些损伤或者可以忍受,或者致细胞于死。6-4PP,6-4嘧啶光产物;8-oxo-G,八氧代鸟嘌呤;CPD,环丁烷嘧啶二聚体;IR,电离辐射;Me,甲基;SSB,单链断裂;TDP,酪氨酸DNA磷酸二酯酶。
求生与自尽中的DNA损伤响应
DNA加合物的毒性主要是由于它阻断了DNA复制或转录时的聚合酶的进程(图一b)。 由简单的烷基化试剂致成的某些DNA加合物(诸如N3-甲基腺嘌呤),由UV辐射导致的光产物,化学武器(如芥子气)和基因毒性抗癌药(例如顺铂、环磷酰胺和美法仑)导致的DNA链内和链间交联,由多环芳烃、杂环胺、黄曲霉毒素和氯乙烯等导致的大加合物[12,13],以及拓扑异构酶抑制剂造成的DNA断裂能通过空间阻碍直接影响DNA和RNA聚合酶的进程。诸如O6烷基鸟嘌呤和N7烷基鸟嘌呤等小加合物,如果它们对应的修复通路(错配修复(MMR)和碱基切除修复(BER))在DNA合成时进行,就会间接阻断DNA聚合酶的进程[14,15]。此等DNA聚合酶之阻滞事件会起始DDR之传信,以稳定被阻滞的复制叉,也会引起细胞周期停滞和DNA修复,直到损伤被修复后DNA复制才会重新启动。如复制重启失败,过长的聚合酶阻滞会导致复制叉的崩塌,并形成一个具有潜在毒性的只有一个末端的DNA双链损伤(DSB)[16],这个DSB是一个无另外DNA末端可连的末端。虽然DSB是DNA损伤所致的主要致毒事件,在肿瘤中,如果自尽通路被沉默,这些损伤就可成为基因组变异形成之基础[17]。DSB形成之后,细胞命运之决定是在DDR激活之后做出的[18]。一个中心问题是,是否DNA加合物自身可激活细胞死亡通路,或者DSB是否为主要凋亡制造者。实验结果说明DSB是一种凋亡的强激活子[19],大多数DNA碱基损伤(例如ROS、活性氮(RNS)和简单烷基化试剂致成的小加合物)导致细胞坏死[20]。坏死的诱导是在修复(诸如BER对N甲基嘌呤和八氧代鸟嘌呤(8-oxo-guanine))过程中聚腺苷核糖聚合酶一(PARP1)被激活之后发生的。PARP1的激活会将细胞内NAD+和ATP池耗尽,从而导致细胞坏死性死亡(也被名为枯死(parthanatos))[21]。顺便说一下,枯死,像凋亡一样被认为是一种保护机制,它可在小DNA加合物修复需求超过细胞修复能力时,抵制细胞之癌变[22]。需要注意的是非同源末端连接(NHEJ)、同源重组(HR)、MMR、BER、核酸切除修复(NER)和蛋白偶联DNA断裂(PDB)对DNA的修复与DDR传信和损伤忍受机制[23-25]是细胞存活的关键通路(图一b)。然而,细胞死亡程序的启动不仅仅因为修复能力的缺失,也更因为与DDR相关的一系列分子互作制止了细胞修复并刺激细胞死亡。
为了让细胞对严重的DNA损伤产生反应,损伤首先需要被检测到,特别是当出现了DSB或复制叉停滞事件。现在已经知道有三个互联的感知系统可以在一个DSB形成后的分分钟内检测到它[26]。在DDR里,那些即时的早期感知子是PI3K相关激酶(PIKK):共济失调毛细血管扩张突变蛋白(ATM)、共济失调和Rad3相关蛋白(ATR)和DNA依赖蛋白激酶(DNA-PK)。从这些感知子开始,通过一些列激酶反应,DDR级联效应开始活化,靶向那些处在求生或自尽的关键节点的双功能蛋白。这些蛋白一方面使下游检验点激活以阻止细胞周期进程,另一方面通过在不同细胞周期背景下刺激NHEJ或HR(例如BRCA1和范可尼贫血互补组丁二蛋白(FANCD2)),募集DNA修复蛋白以促进DSB的修复。以其促进修复和阻止细胞周期的能力,ATM和ATR无疑是求生因子,因为如果敲除它们的细胞系对DNA损伤所致的DSB格外敏感[27]。因此,人们可能会估计ATM和ATR的显性负向突变(一个等位基因为野生型,另外一个为突变型,突变型的存在影响野生型之功能的发挥,译者注)在癌症中应该很少。然而,与预期相反,肿瘤中ATM常常会发生突变[28],伴随这种突变而来的是肿瘤对(基因毒性)治疗获得了抵抗性[29]。这是因为,在DSB水平非常高的时候,ATM和ATR 或者借助p53依赖的[31]和半胱氨酸蛋白酶二(caspase 2)依赖的[32]凋亡通路(图二)的激活,或者在特定的细胞类型中通过E2F1、p73和CHK1(见综述[33])的激活[34]促进细胞的死亡,因此ATM的缺失会导致肿瘤对细胞致死通路具有抵抗性。
�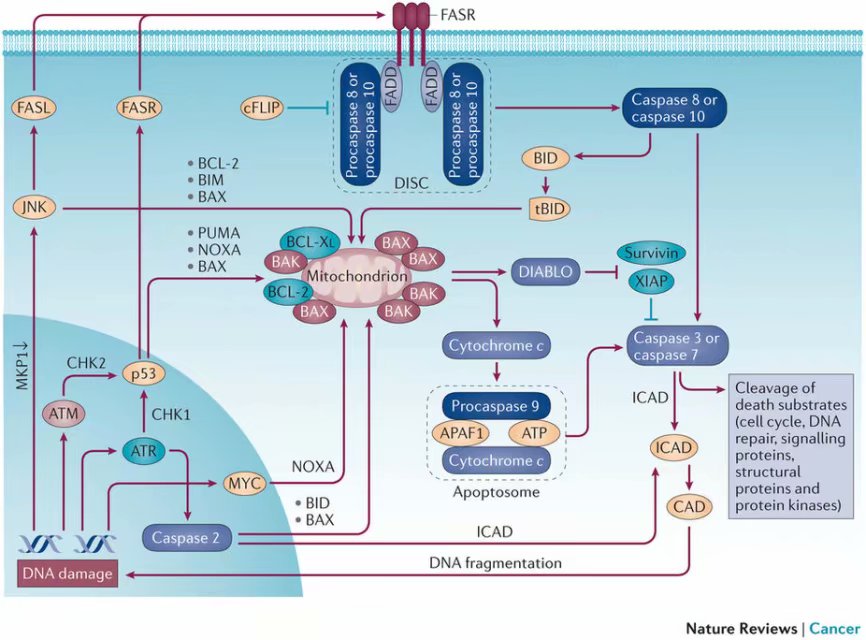 �
�
图二:DNA损伤依赖的凋亡。DNA损伤既可以激活外源的(死亡受体),亦可以激活内源(线粒体依赖的)凋亡通路。DNA损伤响应的促凋亡组分是为JNK、p53、caspase 2,可能还有MYC。这些蛋白激活促凋亡蛋白Fas配体(FASL)、Fas受体(FASR)、BCL-2互作调控子BIM(或名为BCL2L11)、BAX、p53上调的凋亡调控子PUMA(或名为BBC3)、NOXA(或名为PMAIP1)和BH3死亡结构域互作蛋白BID;并下调抗凋亡蛋白BCL-2。这又导致caspase们的蛋白酶活性和caspase激活的DNA酶(CAD,可断裂DNA)的DNA酶活性的活化。DNA损伤也可能抑制了MAPK磷酸酶第一MKP1的转录,MKP1转录的下调导致JNK磷酸化的增加,以及激活蛋白第一(AP-1)活性的增加,最终驱动FASL和外源凋亡途径。APAF1,凋亡肽酶激活蛋白第一;ATM,共济失调毛细血管曲张突变蛋白;ATR,共济失调毛细血管曲张和Rad3相关蛋白;cFLIP,细胞的类FLICE抑制蛋白(或名为CFLAR);DISC,死亡诱导传信复合体;FADD,Fas关联结构域;ICAD,caspase激活的DNase之抑制子;tBID,截断的BID;XIAP,X染色体连锁的凋亡抑制子。
患有由ATM失活突变导致的共济失调毛细血管扩张的病人,罹患乳腺癌的风险大幅增加[35],罹患结肠癌的风险中等程度的增加[36];而ATR功能受损的患者,生出恶性肿瘤的风险也会增加[37]。虽然在肿瘤中ATM突变的频率似乎高于ATR,但ATM和ATR,或通过促进DNA修复,或通过通告细胞死亡[39],都是受损细胞维持基因组稳定性所需要的。然而,这些患者的肿瘤中含有突变的ATM或突变的ATR,却没有死,这说明细胞维持存活的能力超过了这些激酶识别和通告下游因子修复DSB和稳固停滞的复制叉的能力。一种解释是,细胞中还有很多可以解决复制叉阻滞和DSB修复问题,所以在ATM和ATR突变的细胞中DSB不会不修复[40],只是它们可能没有被忠实的修复,而这就导致了基因组不稳定性。一种假说这样猜测:在停滞的复制叉处,ATM和ATR介导的重组修复通过增殖细胞核抗原蛋白(PCNA)的泛素化而与跨损伤合成通路(translesion synthesis,TLS,有致错倾向)发生竞争,TLS可以防止复制叉崩塌,使复制跨过损伤,而细胞也得以存活。ATM和ATR可以通过激活自尽因子(见下文),诸如磷酸化p53的Ser46,而将对死生的均衡切换到自尽的方向。因此,死生的控制不是严格分开的,而是依附于具有反向和正向反馈调节的复杂网络之中的。
求生与自尽之策略
求生策略包含如下机制:各类组成性的或诱导性的修复通路的激活,将致毒损伤修复(图一a);通过TLS跨过致毒损伤;自噬;大限;利用诸如保生素(survivin)、凋亡抑制子(IAP)和X染色体连锁凋亡抑制子蛋白(XIAP)等抗凋亡因子阻止凋亡(图二);激活调控往生基因表达的转录因子,诸如核因子卡波乙(NF-κB)和AKT(又名PKB)(知识框一)。细胞自尽策略包括:凋亡(图二);受控的坏死(枯死(parthanatos),病死(necroptosis)和锈死(ferroptosis))[42];DNA损伤后检验点适应[43];和有丝分裂死[44]。关于这些求生和自尽的通路,以及它们的互作,将在下文详述。
知识框一: 活化调控求生通路的转录因子
¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥
有很多转录因子参与求生机制的调节。我们在此讨论两种:核因子κB(NFκB)和AKT。
NFκB的求生作用
越来越多的证据显示DNA双链断裂可激活NFkB[186]。对于ATM/ATR-CHK1/2-p53轴激活NFkB中,DSB通过PIDD1/RIPK1复合体苏木化NEMO,导致NFkB的重要调控子NEMO的入核[187,188]。因此RIPK1介导的NEMO苏木化对依赖DNA损伤的NFkB激活是极为重要的[189]。DSB激活的ATM也可以与NEMO组成复合体并将之磷酸化[187]。ATM-NEMO复合体被转运至细胞质[190],结合NFkB激酶抑制子IKK复合体,IKK复将IkB磷酸化,导致NFkB下游求生靶基因的转录。在这些NFkB转录的基因中,有BCL2L1、BCL2A1[191]、IAP2[192]、TRAF1和TRAF2[193](如图)。因此NFkB激活的转录,既抑制线粒体介导的,又抑制死亡受体(TNFR)介导的凋亡。此外,NFkB也上调MAPK磷酸酶第一(MKP1)[194],来抵抗持续DNA损伤所致的JNK的磷酸化和JNK驱动的凋亡(图二)。其后,在DNA损伤试剂(如拓扑异构酶第一抑制剂)处理时抑制NFkB的激活导致凋亡的加强和体内肿瘤生长的减缓[195]。
AKT的求生作用
AKT激酶以PI3K依赖的方式[196]磷酸化、因而抑制促凋亡蛋白,或者激活抗凋亡之系统。在这之中,AKT磷酸化BAD[120]、ASK1[151]、caspase 9[152]和MDM2[153],因此抑制凋亡(如图)。AKT还可促进IkB的降解,以此激活NFkB,NFkB促进抗凋亡基因的转录来抑制凋亡(如图)。
因DNA损伤试剂之别,AKT可以被DNA-PK[146]、ATM[147]、ATR[148]亦或MRE11[149]激活。Ser472磷酸化的AKT与磷酸化的ATM一起定位于DNA损伤导致的γH2AX集落[149]上,此种共定位可被E3泛素连接酶RNF168促进。AKT还在DDR激活时影响p53的稳定性,因为AKT可能通过与MDM2的反应,作用于p53的降解[146]。AKT对凋亡的抑制,增加了DNA损伤细胞的存活力,这种机制可见于很多癌细胞[197]。
除了上述AKT之功用,AKT还可直接促进DNA修复。在这之中,DNA损伤后,AKT结合DNA-PK,将之磷酸化,调控其在损伤位点的募集,襄助DSB的NHEJ接合的效率[198]。因此,AKT及调控DSB修复和求生;反过来,也调控细胞死亡相关的依赖p53的和不依赖p53的响应(如图)。
� �
�
¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥¥
DNA损伤后p53在均衡生与死中的作用。不同水平的损伤可能引发不同的响应;这就是说低水平的DNA损伤引发修复和求生机制而高水平损伤致使细胞死亡。因为p53具有靶向求生基因和自尽基因的功能[39](图二),因此推测p53处在生死抉择的中心是合理的。细胞怎样定量DNA损伤,相关策略如何产生?理论模型推测,p53的不同活化程度依赖于不同的DNA损伤程度,而这一转变受复杂的正向(ATM、ATR-CHK1/CHK2-p53)和反向(MDM2)反馈环路的控制[45-47]。简单地说,低水平的DNA损伤只短暂的激活了p53,而高水平的DNA损伤则导致长久的p53激活。因为p53结合那些与阻止细胞周期相关基因(例如编码p21的CDKN1A)的启动子的亲和性比较高,而它结合那些与凋亡相关基因(例如p53诱发基因三(PIG3,或TP53I3))的启动子的亲和性比较低[48],因此不同稳定程度的p53可能造成其靶基因的差异表达。p53对启动子的选择性也依赖于RNA和染色质互作因子。因此,在诱导敲除小鼠模型里,正常细胞中Cdkn1a的表达依赖于RNA解旋酶p68(也被称为DDX5)[49],而在肺癌细胞和乳腺癌细胞中PIG3和p53AIP1(p53调节的凋亡诱发蛋白一)的表达依赖于CAS(细胞凋亡敏感性蛋白,也被名为EXP2和CSE1L)[50]。进一步的,p53与ASPP1(p53刺激凋亡蛋白一)和ASPP2的直接互作[51]诱发了凋亡,而p53与ASPP蛋白抑制子(iASPP)[52]的互作则可防止凋亡。诸如NF-κB(知识框一)等求生通路与p53之间的对话也影响了p53的求生和自尽两种角色的转变。已知的是,对p53及其下游作用的调控是复杂的,而且是细胞类型和癌症类型特异的。
除了上述机制,p53从“制止者”和“修复者”向“刽子手”的功能转换还依赖其氮末端的磷酸化和碳末端的乙酰化[53](图三)。在诸多磷酸化中,由ATM、ATR、CHK1、CHK2和DNA-PK介导的Ser15、Ser37、Thr18和Ser20的磷酸化,将p53从它的抑制性结合伴侣MDM2上解离,并协助其进入细胞核发挥转录激活作用,使p53施展它的制止者作用,例如,诱导CDKN1A基因的表达[54]。而p53的Ser46的磷酸化与p53刽子手的角色特异相关[55],据推测,在响应化学和物理胁迫之时,这一磷酸化可以p53的启动子选择性改变,并帮助p53诱导诸如NOXA(也被称为PMAIP1)[56]、PTEN[57]和TP53AIP1[58]等凋亡基因的表达,以至于细胞凋亡。有很多候选激酶被发现可与p53互作并将其Ser46磷酸化,诸如同源框互作蛋白激酶二(HIPK2)[59],p38[60],蛋白激酶C嘚塔(PKCδ)[61],p53依赖损伤诱导核蛋白一(p53DINP1;也被称为TP53INP1)[62]以及双特异酪氨酸磷酸化调控激酶二(DYRK2)[63]。TM和ATR被认为作用于p53之Ser46磷酸化的上游(可能是通过XIAP相关因子一(XAF1)),它们可将E3泛素连接酶七缺同源蛋白一(SIAH1)的Ser19磷酸化。这一修饰将HIPK2-SIAH1复合体解聚,以此稳定HIPK2蛋白[65],使之与核内PML体的早幼粒细胞白血病蛋白(PML,一个有效的促凋亡肿瘤抑制子)、p300、CREB结合蛋白(CBP,也被称为CREBBP)和p53结合,其最终效果是上调p53的靶基因[66]。
�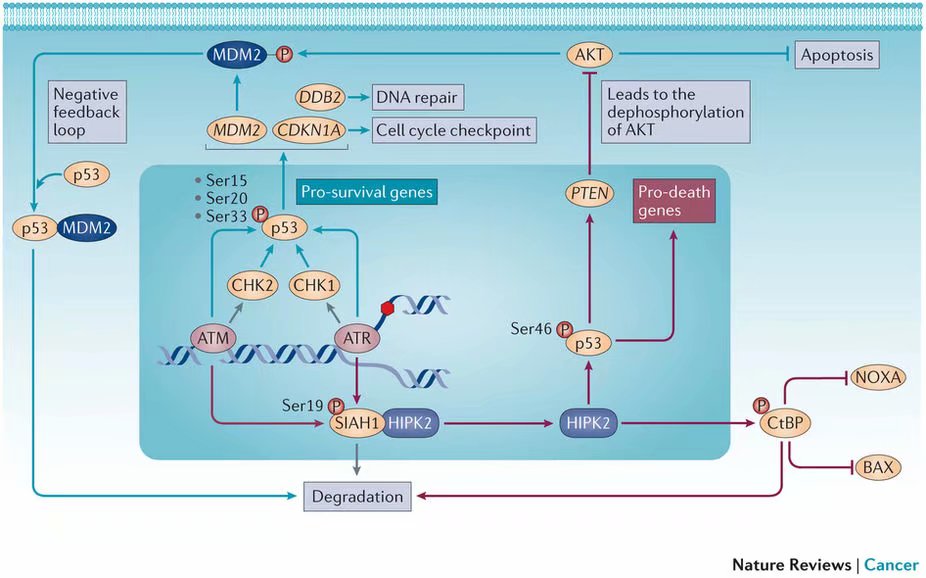 �
�
图三:DNA损伤既激活了求生信号,又激活了寻死信号。求生信号:ATM和ATR磷酸化p53,因而导致CDKN1A(周期蛋白依赖的激酶抑制子第一之甲)、DDB2(DNA结合蛋白第二)和MDM2(一种E3泛素连接酶)等求生基因的表达。这是一个DNA损伤早期的响应,它是检验点激活和DNA修复所需的。寻死信号:ATM和ATR导致HIPK2(同源结构域互作蛋白激酶第二),HIPK2复又磷酸化p53之46位丝氨酸,p53之Ser46磷酸化导致促凋亡基因之表达。HIPK2也引起CtBP(碳末端结合蛋白)的降解,导致促凋亡蛋白NOXA和BAX的激活。这可能是DNA损伤晚期的响应,如果DNA修复失败了的话。AIAH1,七伤同源第一(E3连接酶)。
顺便说一下,p53(在Ser46和其它位点)的去磷酸化被认为是依赖p53诱导磷酸酶WIP1(也被称为PPM1D)的[67]。因此,p53(Ser46)磷酸化和去磷酸化的负反馈环路可能存在,而HIPK2稳定化的失衡(可能是被高激活亦或持续激活的ATM亦或ATR(在高水平损伤的情况下)启动),可将细胞由修复状态切换为凋亡状态。尽管如此,确知的是,p53是被异常复杂的调控的,需要更深入研究其机制以探索DNA损伤后细胞如何决定求生还是寻死。
在癌生物学中,癌细胞的凋亡可能因为E3泛素连接酶SIAH1的活性上调而被关闭。SIAH1与低氧诱导因子(HIF)一起,参与细胞的缺氧反应[68]。有趣的是,在神经胶质瘤发生过程中,异柠檬酸脱氢酶一( isocitrate dehydrogenase 1 ,IDH1)的突变可能与HIF1的活性相关[69]。这使人很容易推测,由于HIF活性增高导致的SIAH1介导的HIPK2的降解,使得坚硬缺氧的肿瘤对化疗诱导的细胞死亡产生了抵抗。如同WIP1过表达所观察到的结果,可能这里谈到的效应子大多与癌症的发生相关[70]。当然,促凋亡p53靶基因的转录还需要超越由抗凋亡蛋白BCL-2[71]、保生素(survivin)和XIAP[72]设定的门限,以激活细胞求死通路(图二)。具有高水平抗凋亡蛋白的肿瘤细胞,其化疗后的凋亡响应或者更低或者无有[71,73]。因此,靶向肿瘤细胞的E3连接酶SIAH来辅助癌症治疗可能是有益的尝试[74]。
转录组损伤与细胞死亡
由DNA损伤导致的细胞死亡,除了单单考虑DNA损伤本身,思考在此过程中的转录组的稳定性、可诱导性和其调控也是很重要的。一方面,在转录过程中抑制RNA聚合酶II会诱导凋亡;但在另一方面,求生存和抗生存因子在癌症发生时由表观遗传介导的下调也影响细胞的死亡[76]。
首先,对于RNA聚合酶II的抑制的模型,通常的,求生因子是从大基因(大小有许多千的碱基长)转录而来的,而寻死因子由小基因(几千碱基)转录[77]。因此,抑制转录的DNA加合物可能更倾向于抑制求生基因的转录,因为大基因比小基因更有可能成为损伤的靶点。于是,未修复的转录阻断性加合物阻断了求生因子的转录,将生死平衡的抉择推向了寻死。
这一机制可以使生死平衡倾斜,其中转录抑制既可以通过ATM亦或ATR介导的信号传导[78],也可以通过MDM2转录的抑制[79]间接的造成p53的磷酸化和积累(图三)。相同的转录阻断性DNA损伤也可以阻断由p53调控的求生基因的转录,诸如损伤特异的DNA结合蛋白(DDB2)和干性皮肤色素沉着互补组丙( xeroderma pigmentosum complementation group C,XPC)[80-82],以此影响生死平衡。
大块DNA加合物是强效的转录抑制剂。这些加合物可以在RNA聚合酶停滞后,被伴转录核酸外切修复(TC-NER)[83]修复。如果TC-NER未能移除大块加合物,Ser15位点的p53磷酸化会积累[84]。p53的Ser46有没有被磷酸化还不清楚。在癌生物学中,DNA损伤依赖的转录抑制激活p53诱导的凋亡可能极为重要,因为可以想见,高效转录可能会增进癌细胞的增殖(和代谢)。
DNA损伤与转录互作的另一实例来源于对转录因子FOS缺陷细胞的研究。在UV辐射与大块DNA损伤诱导试剂处理之后,FOS会在即时早期[85]和晚期[86]响应(immediate-earlyand late)中被迅速诱导。FOS缺陷细胞的标志是其对导致大块DNA损伤形成的基因毒剂(比如UV辐射和苯并[a]芘( benzo(a)pyrene)[87,88])的高度敏感性,这种DNA损伤与重度DNA复制和转录的阻断相关[89]。DNA加合物的修复在FOS缺陷细胞中很是缓慢,此乃是FOS激活蛋白一(AP-1)调控的NER蛋白XPF和XPG的转录失败引起的[89]。即时早期(immediate-early)FOS的诱导促进XPF和XPG的转录,这是很重要的,因为它维持了细胞的NER的能力。这一早期响应被认识可以持续调节这些不稳定的NER基因转录物并可补偿它们的短缺[90]。XPF和XPG都是内切酶,它们可切断DNA的5’和3’而形成DNA断裂[91],因此它们在细胞中维持在很低的水平是可以理解的,因为细胞需要避免不想要的基因组损伤。依赖于基因毒剂胁迫水平,对这些修复基因做出的精细(稳定状态的)上调,对维持基因组稳定性明显是重要的。值得注意的是,对系统施加低剂量的DNA损伤毒剂,可以防止后续大剂量毒剂造成的细胞死亡[90]。这暗示晚期FOS激活,使细胞激活了求生的通路[86]。因此,生死之抉择可能因急性和慢性基因毒剂处理的不同而不同。
DNA损伤后的FOS激活是一种普遍的胁迫响应,这种胁迫响应涉及JNK和p38激酶的激活,而这些激酶的激活又增加了AP-1的活性。NER缺陷突变会造成JNK和p38激酶的持续激活,说明DNA损伤会引起胁迫响应[92]。AP-1的长时间激活会增加Fas配体(FASL,或名为FASLG)的表达[93],因而推动了外源凋亡通路(图二)。
概括而言,NER过程所致的DNA损伤,通常具有细胞毒性,如其不治,会导致JNK和p38激酶活性的持续上调(求死开关)[92],这种上调可以通过MAPK磷酸酶第一(MKP1,或名为DUSP1)的下调引起(可能是MKP1转录阻滞的结果),而MKP1可将JNK和p38灭活[94](图二)。这就会导致之后的JUN-ATF2控制的FASL的上调[95]。在这一事件中,DNA损伤后细胞是生是死,决定于修复蛋白XPF和XPG的合成、MKP1下调的程度、JNK和p38激酶活化时间的长短、诱导FASL的程度以及FAS(又称为CD95和APO1)死亡受体系统的效果(FAS死亡受体系统在诸如黑色素瘤[96]等肿瘤中通常处于沉默状态)。在此事件中,DNA损伤导致了FOS的上调和XPF-XPG介导的NER修复,其中,对于癌细胞,FOS的含量可能起到决定作用。对这一系统的控制可能蕴含一种可行的癌症治疗方法。
考虑细胞对DNA损伤诱导剂之响应时,也许关注表观修饰改变所致之转录组变化,因为表观遗传也可调节DNA修复,以及死亡通路的激活和执行。例如,在肿瘤中,特别是神经胶质瘤,DNA修复基因第六位氧甲基鸟苷酸DNA甲基转移酶(MGMT)通常会因其启动子区域的高度甲基化而被沉默[97]。MGMT可将鸟苷酸第六位氧上面的烷基加合物移除,因此可阻止导致DDR激活和细胞死亡的事件发生。因此,正因为神经胶质瘤的MGMT是沉默的,因此它可以相应烷基化化疗法[98]。凋亡所需因子的沉默在其它癌细胞中也有过报道。上面已经说过,DNA损伤不治所致的p53的长时间稳定化,会引发凋亡。p53的一个关键靶基因就是高甲基化于癌症第一(HIC1)基因,它编码司徒林第一(SIRT1)的转录抑制子。SIRT1可将p53去乙酰化,从而负向调节p53的转录活性[99]。因此在细胞中常见的HIC1的表观遗传沉默,消除了SIRT1的转录抑制,导致p53的提早失活,使得p53引发凋亡的通路受损[100]。除此之外,直接执行凋亡过程的因子也有一些在癌细胞中被表观沉默。例如,黑色素瘤具有沉默的凋亡肽活化因子第一(APAF1)[101]和半胱氨酸蛋白酶第八( caspase 8)[96]的启动子,使得这种癌细胞对基因毒疗法具有抵抗性。
DNA损伤导致的大限
DNA损伤是致细胞于其大限的诱导者[102]。当前对于大限的认知源自于如下发现:人二倍体成纤维细胞(HDF)只能经历有限的细胞分裂(复制大限),以及暴露于高剂量的诸如电离辐射或丝裂霉素C(MMC)的HDF细胞虽然不在增殖,但代谢活动却还照旧[103]。这种DNA损伤致细胞于大限的现象,目前被用来制作饲养层细胞[103]。
有证据表明,导致细胞大限的DNA损伤信号源自端粒上的DSB,此种DSB导致了持续的DDR信号[102]因此激活了p53依赖的[104]或p16(也被称为INK4A)依赖的[105]细胞周期阻滞(图四)。一个浅显的问题是,为什么基因组区域的DSB只能短暂的激活DDR,而端粒区的DSB则会产生持续的DDR信号。此问题可以通过下面这一观察解答:端粒内或端粒附近的DSB未被修复。端粒重复结合因子第二(TRF2)是鞘素(shelterin)的一个组分,它在体外可结合端粒的双链TTAGGG重复;在体内定位于全部端粒区域[106],并通过阻断连接酶四(ligaseIV)向端粒内或端粒旁之DSB位点之募集,而抑制NHEJ介导的DSB修复的完成[107](图四)。以此,由于端粒的DSB是持续的,其DDR响应也是持续的,细胞也就进入了一种分裂静止的状态;而基因组区域的DSB可以被修复,因此其DDR所致细胞周期阻滞也是暂时性的。TRF2以此使人类端粒免于末端融合[108],而末端融合则可能导致恶性癌变[109]。这也解释了为何产生存活而不增殖的HDF需要大剂量的DNA损伤制剂(例如电离辐射大于70格雷),因为DSB发生在端粒区的概率要小于其发生在基因组区的概率。值得注意的是p53是这一通路活化所需要的,这可以解释为什么HDF在大剂量DNA损伤制剂处理后不会死亡,而RAS活化或p53突变的细胞则会走向凋亡而不是走向大限[110]。
�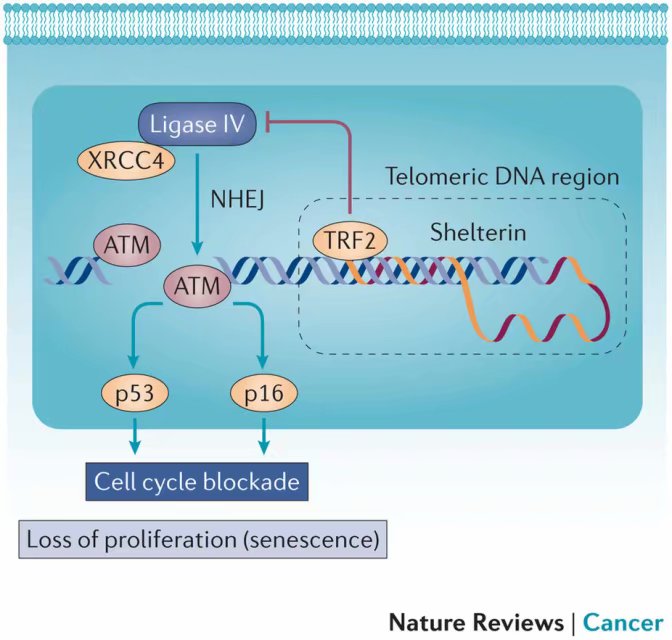 �
�
图四:依赖DNA损伤的大限。在端粒上,鞘素蛋白端粒重复结合因子第二(TRF2)通过抑制连接酶第四,妨碍了DNA双链断裂的非同源末端连接(NHEJ)修复。这导致持续的ATM向p53亦或p16的信号传递,以及不可逆的细胞周期阻滞。
值得注意的是,中等程度的化学基因毒剂可导致大限。因此,用于治疗神经胶质瘤的将底物甲基化的试剂,替莫唑胺( temozolomide ),可以在低的临床浓度下导致一小部分细胞的ATM和ATR激活和大限[111]。由此看来,端粒未修复的DSB可能不是DNA损伤细胞至于大限的唯一机制。因此,尚需解决的复杂问题还包括:低剂量的基因毒剂如何诱发非端粒模式的大限,这些细胞是否可以从沉默状态恢复到增殖状态?如果细胞可重新增殖,那么这一机制的研究意义是重大的,因为肿瘤细胞可以在治疗时进入非增殖状态,并在肿瘤治疗后完全恢复增殖。
自噬之于DNA损伤所致之生死抉择
DNA损伤可在三个水平调控自噬。DNA损伤可防止mTOR1对自噬的抑制,并激活ATG1(也被称为ULK1)和Beclin1复合体。这种作用涉及到PARP1、JNK、ATM和p53等诸多信号通路(图五)。
� �
�
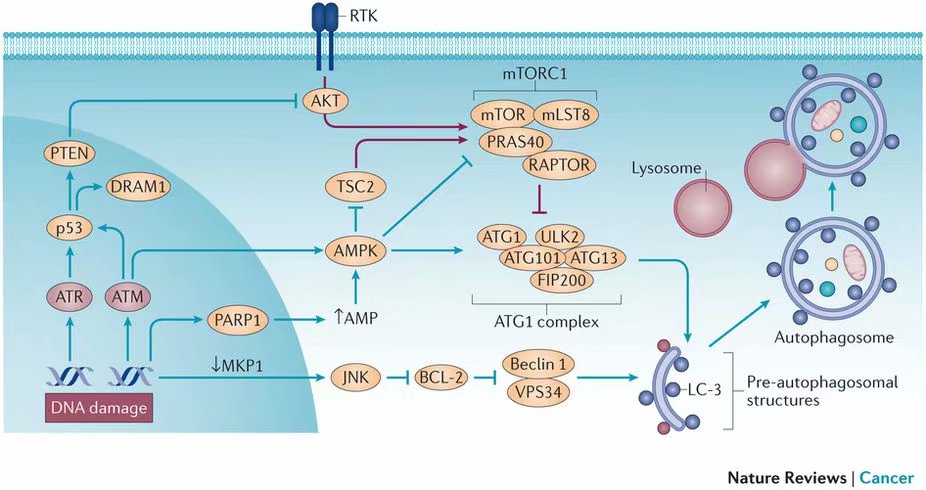
图五:依赖DNA损伤的自噬。在理想的生长条件下,受体酪氨酸激酶(RTK)通过PI3K-AKT在通过mTOR复合体第一(mTORC1)来抑制自噬。mTORC1包含mTOR、MLSTUbL8、PRAS40(或名为AKT1S1)和mTOR调控关联蛋白RAPTOR。AKT直接磷酸化PRAS40,或者通过马铃薯球蛋白(tuberin,TSC2,在Ser939,Ser1086/Ser088和Thread422)的磷酸化间接磷酸化PRAS40,来抑制自噬。ATG1复合体,包含自噬相关蛋白第一ATG1(或名为ULK1)和ULK2、ATG13和FIP200(又名为RB1CC1),是自噬的主要启动者。mTORC1通过RAPTOR与ATG1-ULK2之结合,以及mTOR对ATG13和ATG1-ULK2的磷酸化来抑制ATG1复合体。ATG1-ULK2的磷酸化抑制了其激酶活性,因为ATG1-ULK2-ATG13-FIP200激酶活性是自噬所需的,因此之故,自噬启动就被抑制住了。与AKT和mTORC1对自噬的抑制相反,自噬可以被能量敏感激酶AMPK激活。如示,AMPK被高水平AMP激活,被高水平ATP灭活。当AMPK被激活后,它可磷酸化RAPTOR和TSC2(在Thr1227和Ser1345),因而移除mTORC1对自噬的抑制。AMPK进一步磷酸化ATG1,因而强化ATG1之活性而促进自噬。活化的ATG1-ULK2磷酸化FIP200和ATG13,这有导致ATG1复合体移动到自噬前体膜上,启动自噬。经典的自噬前体膜结构的启动时Beclin 1和VPS34组成的复合体来启动的,其中VPS34又名为PIK3C3,是第三类PI3K。DNA损伤是通过灭活mTORC1,激活ATG1和Beclin 1复合体激活自噬的。ATM,共济失调血管曲张突变蛋白;ATR,共济失调血管曲张和Rad3相关蛋白;DRAM,损伤调控的自噬调控蛋白;JNK,JUN氮末端激酶;MKP1,MAPK磷酸酶第一;PARP1,聚二磷腺苷核糖聚合酶第一。
自噬之时,p53具有很多功用,这些功用是通过转录激活PTEN[112]和DRAM1(DNA损伤调节的自噬调解蛋白第一)[113]实现的。p53转录激活PTEN,可以抵消受体酪氨酸激酶(RTKs)通过阻止AKT活化对自噬的抑制。因此,细胞中存在两种相反的势,一种是生长因子激活AKT[114],一种是DNA损伤依赖的通过PTEN实现的对AKT的抑制[115]。DRAM1对自噬起始似乎没有功用。DRAM1的三个剪接变体(SV1[113],SV4和SV5[116])定位于溶酶体或内体(SV1),或过氧化物酶体和自噬体(SV4和SV5)。由于这些细胞器都与自噬之起始无关,因此p53诱导的DRAM1之表达可能是为细胞应对DNA损伤所致之自噬做准备。
PARP1是一种染色质相关酶,它涉及到有烷基化试剂和ROS等物致成的小块加合物的碱基外切修复[117]。它可通过聚二磷腺苷核糖基化(poly(ADP-ribosyl)ation)修饰很多蛋白,此过程会消耗大量NAD+。用多柔比星( doxorubicin,拓扑异构酶II的抑制剂,也可导致氧化胁迫[118] )处理PARP1野生型细胞,可通过ATP和NAD+的减少,间接激活自噬。当PARP1被敲除后,多柔比星就不再产生强的自噬响应了[119]。如果PARP1被抑制或去除,饥饿所致自噬的发生会延迟[120]。饥饿会增加细胞内ROS水平[121],ROS所致的DNA损伤激活PARP1,导致能量耗损。因此,在DNA损伤修复时PARP1的激活,是适当诱导自噬的之先决条件[120]。所有的形成ROS的DNA损伤试剂,诸如UV-A,皆可致成自噬[122]。因此,DNA损伤可能通过PARP1与自噬紧密相连。
DDR信号可以直接的激活自噬。其中ATM可激活AMPK,AMPK又激活自噬[123]。不需要PARP1的DNA损伤修复亦可以引发自噬。其中一个例子是,处理O6MeG时生成的DSB[124]可以激活ATR和ATM[125]。O6MeG致成的自噬是依赖于ATM的[111]。电离辐射的应用扩展了我们对ATM在自噬中的作用的理解。电离辐射所致的DSB可激活ATM,ATM又通过丝苏氨酸蛋白激酶第十一(STK11)和AMPK代谢通路激活肿瘤抑制子 马铃薯球蛋白 (TSC2),以此抑制自噬抑制子mTORC1[126,127]并激活ATG1复合体。值得注意的是,伴侣蛋白介导的自噬导致活化CHK1的降解,因而作用于DDR的负反馈调节[128]。
白克林第一复合体(Beclin 1 complex)可以被BCL-2抑制[129]。BCL-2是JNK下游的磷酸化底物[130],磷酸化后BCL-2不在抑制白克林第一复合体,自噬前体膜就可以形成。如上所述,MKP1抑制JNK,因此转录阻滞性DNA损伤所致MKP1的下调将致成JNK之持续性活化。因此可以预计,所有转录阻滞性DNA损伤,超过某一门限,就可能激活白克林第一复合体(图五)。
在DNA损伤程度低时,自噬是一中求生机制。而DNA损伤程度高时,自噬就变成不受控制的过程,生死之抉择将向自尽推进。在分子水平上,持续性之DNA损伤导致ATM的长时间活化,由它抑制的mTORC1和激活的自噬[127]就不会改变[131]。在此种情形下,过度的自噬导致细胞自我蚕食以至于死。在此事件中,ATM可以被FOXO3活化[132],FOXO3通常结合于DNA。DNA损伤时,FOXO3从DNA上解离,与ATM作用,致使ATM/ATR-CHK1/2-p53轴线的持续性活化。此外,FOXO3还调节LC3(又称作MAP1LC3A)和BCL-2/BNIP3的转录,这两者是自噬所需的[132]。因为既可激活p53依赖的细胞死亡,又可激活自噬通路,因此FOXO3可能处于求生与寻死抉择的中间位置。研究表明三氧化二砷(As2O3)所致之恶性细胞死亡,是通过自噬完成的[133]。三氧化二砷影响PML体之功用[59],而PML体可通过HIPK2磷酸化p53之Ser46。因此,可能存在这样一种联系,它以PML为中心,连接自噬所致细胞死亡和p53之Ser46磷酸化。虽然自噬所致之生,或为形成肿瘤之机制[135],或可作为治疗肿瘤之靶点[136],但自噬所致之细胞死亡,可能对抑制肿瘤有关键作用[137],并可能影响癌症治疗的效果。
求生与求死之间之互戏
基因毒剂所致之自噬、坏死与凋亡之间的互感是复杂的,目前对此认识还很不足。上文谈到其中的几个方面,下面我们将关注涉及这一复杂度的一些事例。概括而言,DNA损伤,通过ATM/ATR-CHK1/2-p53轴,激活了细胞周期检验点,阻止细胞周期进程,并激活了DNA的修复。DNA损伤问题解决完后,ATM/ATR-CHK1/2-p53级联反应因去磷酸化而被关闭[67],因此坏死或凋亡所致之细胞死亡就不会发生。细而论之,DDR通常通过信号蛋白和相关通路,同时激活诸如凋亡[138]和坏死[21]之类的求死机制,以及包括DNA修复[90]和自噬[111]的求生机制,这就使问题变得复杂起来。在这一过程中,各类门限发挥着重要作用。
细胞死亡之门限通过诸如凋亡等下游求死通路来实现。主要的死亡调节子包括保生素(survivin)、IAP和XIAP,这三者联合起来可以抑制半胱氨酸蛋白酶(caspases)[72,139],因此阻止凋亡所致的细胞死亡。保生素不仅在胚胎和胎儿发育中的高速增殖的组织中高表达[140],在肿瘤中也高表达[141]。保生素敲除细胞对DNA损伤所致之凋亡敏感,因为激活凋亡的门限降低了[142]。在胚胎发育中,组织中的细胞在持续不断的进行细胞分裂。内源DNA损伤所致的复制压力[143]有可能推动这些细胞进入凋亡状态。而保生素可以通过抑制半胱氨酸蛋白酶抑制凋亡的提前激活,以此减轻复制压力所致的细胞凋亡。在肿瘤中,相同的机制导致肿瘤对抗肿瘤药物产生抵制[144]。因此,为了使凋亡发生,必须激活足够的半胱氨酸蛋白酶,克服由保生素和XIAP等抗凋亡蛋白建立的抑制凋亡的门限。值得关注的是,因为自噬求生之门限依然还不清楚,因此我们说在DNA损伤程度低时,自噬可使细胞免于凋亡,这是合理的。癌细胞除了过表达保生素,也有其它方式来控制激活求死通路之门限。例如,黑色素瘤抑制促凋亡蛋白APAF1[101]和caspase 8[96]之表达;淋巴瘤大量表达诸如BCL-2等抗凋亡蛋白[145]。死亡激活门限的建立可以被信号传导的激活来加强。根据不同的DNA损伤试剂,AKT可以被DNA-PK[146]、ATM[147]、ATR[148]亦或MRE11[149]激活。AKT可通过激活或抑制细胞死亡之BCL-2相关激动剂蛋白BAD[150]、凋亡信号调节激酶第一ASK1(又称作MAP3K5)[151]、caspase9[152]和MDM2[153]来强化这一门限。在DNA损伤持续存在,DDR和凋亡信号不止之时,足够的半胱氨酸蛋白酶被激活,它们克服门限抑制,裂解白克林第一蛋白(Beclin1)[154],因此将后续的自噬启动阻断,不在接受自噬的益处。重要的是,白克林第一的裂解碎片通过与线粒体作用,进一步刺激凋亡。由此,门限抑制被打破,细胞走上凋亡之路。
DNA损伤时,如果细胞激活的半胱氨酸蛋白酶不足以使细胞从自噬切换为凋亡,而DNA损伤又不能被修复,那么细胞将借助可控的坏死来去死。需要关注的是,大多对可控细胞坏死的研究,都是先把系统的凋亡通路抑制住:或者通过小分子抑制剂[155],或者通过敲除[156]或敲低[157]死亡受体和相关结构域。在凋亡被抑制时,DNA损伤细胞倾向于步入可控坏死进程。这与可控坏死在正常情况下的生物功能是一致的。感染包含凋亡抑制蛋白病毒的细胞会用可控坏死为细胞死亡之机制[158]。同此,抵抗凋亡的癌细胞对坏死诱导非常敏感[159]。要知道,凋亡是一依赖能量的过程,而坏死则无需甚多能量[160]。大量碱基损伤或DNA单链损伤所致的过量PARP1激活,会使细胞由凋亡转变为可控坏死[161]。也就是说,控制细胞从凋亡到可控坏死的关键因素有二:第一,细胞内凋亡系统的成效;第二,是否有足够的能量满足凋亡的启动和完成。
在此,我们简要介绍两种可控坏死: 枯死(parthanatos),病死(necroptosis)。病死需要RIPK3的激活[162]。RIPK3可以被肿瘤坏死因子受体信号通路,借助RIPK1激活[163];也可以通过细胞质DNA借助DAI(又称ZBP1和DLM1)来激活[164];又可以通过脂多糖或双链RNA借助类刀受体(Toll-like receptor)和TICAM1之作用来激活[165]。Caspase 8和细胞类FLICE抑制蛋白(cFLIP,或名为CFLAR)[166,167]可抑制RIPK3;因此,诸如IAP1和IAP2[168]等对caspase 8的抑制,可以激活坏死而抑制凋亡。RIPK3与RIPK1、DAI或TICAM1一起,导致MLKL定位到细胞膜[169],最终使得细胞渗透压平衡丧失。
所谓枯死者,乃因PARP1之高度激活所致也[170]。PARP1之超激活使得细胞内NAD+下降[171],引起胤化(氧化磷酸化,氧化的声母,磷酸化的韵母,组合成一个新读音,胤。书写为左羊右粼)[172]和糖酵解[173]生成ATP之灾难事故。枯死是DNA损伤所致的一种最常见的可控坏死。坏死是否是由DNA损伤所致尚难说清。而枯死是由离子辐射[174]、ROS化学诱导剂、以及甲基化和乙基化试剂[21]等基因毒剂导致的。大量的DNA损伤会导致PARP1的超激活,随之就是细胞内NAD+和ATP的缺失。既然ATP既作用于凋亡,又作用于坏死,那么如果说细胞的能量是DNA损伤细胞求生寻死的重要因子,也不为过[175]。与之一致的是,自噬和能量感知子AMPK对于防止DNA损伤引发枯死具有重要作用[176]。DNA损伤形成后,诸如BER等许多耗能过程被激活,细胞内NAD+和ATP水平降低。AMP含量的增加,将激活AMPK,AMPK再通过自噬等机制恢复ATP水平。当DNA损伤程度超过某一极限,细胞再无能力修复它,或者DNA损伤所致的耗能过程将ATP和NAD+(PARP1介导的修复)消耗到AMPK再无能力恢复细胞能量时,细胞就将死掉了。在这之中,AMPK的活化,可以通过自噬来防止DNA损伤所致之细胞死亡,又可通过为凋亡提供充足ATP,而使细胞去凋亡而不是去枯死。
由死到生的变动
凋亡有许多特征,诸如线粒体膜功能障碍、细胞收缩、细胞膜变化以及半胱氨酸蛋白酶激活,这些特征也是凋亡的早期和晚期的标志[177]。如果DNA损伤信号被干扰,凋亡过程能否逆转呢?可能在某一节点是无法逆转的,但这一节点尚未明确界定[178]。对细胞死亡进行干扰的模型是通过用炸思( jasplakinolide )瞬时诱导线粒体损伤和半胱氨酸蛋白酶活化,并通过干扰此处理来研究的。在这之中,当细胞核碎裂发生,凋亡就不可逆转了[179],说明在从生到死的过程中,到了某个点之后,再想生,就不可能了。这一节点可能由高水平的半胱氨酸蛋白酶来决定的,半胱氨酸蛋白酶激活的DNA酶(CAD)是切割基因组DNA所需的。而这可能会被保生素或XIAP等抗胖蛋白酶(Caspase,取半之声母,胱之韵母)之物影响,而保生素和XIAP在癌细胞中是高表达的[73]。凋亡逆转,是为复活(anastasis)可能是一种肿瘤相关过程,因为凋亡DNA断裂不完全,会导致染色质畸变(chromosomalaberrations)和基因组不稳定[179]。DNA损伤所致之凋亡过程,是否可在DNA损伤信号减弱之后停止,是一个值得关注的问题。
结论
夥矣DNA损伤所致之细胞响应。它激活细胞周期检验点,在损伤干扰复制机制之前,给细胞以时间去修复损伤。如修复失败或能力不足,DNA损伤会阻碍复制和转录。DDR会激活下游寻死通路。因此DNA损伤细胞的生存,仰仗于细胞的修复能力、增值水平、DNA损伤程度、p53和关键DDR蛋白(诸如ATM、ATR和DNA-PK)之状态、激活DNA损伤修复基因的成效(依赖于表观修饰和转录因子)以及下游细胞求死通路的执行状况。DNA损伤修复能力因细胞类型之不同而差异巨大,例如:人胚胎干细胞对大多数DNA损伤之修复能力强于分化细胞[180];单核细胞和肌肉细胞缺乏BER[181,182],而某些癌细胞,诸如转移性黑色素瘤(metastaticmelanoma),修复能力则变强[183];对MGMT之修复亦有差异,比如神经胶质瘤[97,98]。一言蔽之,低水平DNA损伤激活DNA修复(以DNA修复基因XPF、XPG、DDB2、XPC、XRCC1和其它基因[90])的表达上调为标志;而当DNA损伤程度过高,修复能力饱和时,未修复的损伤会激活凋亡、可控坏死和自噬所致自损等死亡程序的其中一个。虽然还不清楚这些通路之间如何切换,但p53的磷酸化和抗凋亡门限可能是决定细胞生死的关键节点。ATM和ATR的功能涉及到DNA损伤的各种可能结果。因此,ATM和ATR可能是首要决策者,指导者效应子(例如p53)发挥功能。具有抵抗力的肿瘤携带突变的ATM[29],说明ATM对启动细胞寻死通路是重要的。值得关注的是,p53的抑制,既可以是癌细胞对药物敏感,也可使其对药物抵抗,这决定于所用之基因毒药物。大块加合物激活的p53上调DNA修复和耐受基因(XPC、DDB2和DNA聚合酶η(POLH)),这些基因是一系列求生基因;而小加合物,诸如O6MeG,其上调的凋亡基因将占据上风[138]。虽然很多工作关注不同的p53的修饰如何影响不同求生寻死基因的启动子的激活,但对于为何p53缺陷细胞在DNA损伤后走向死亡还不清楚。也就是说,一种独立于p53的,通过MKP1、JNK、caspase 2和其它可能因子产生的DNA损伤信号通路还需要深入研究。学界已经意识到DDR所致求生因子(诸如DNA损伤和耐受)和细胞死亡所需超越的门限是非常重要的。针对这些靶点的抑制剂正在被研发和用于临床试验(可见附表)。针对ATM/ATR-CHK1/2轴、细胞周期效应子和DNA修复通路的抑制剂,联合基因毒剂化疗可能是管用的,但它也可能给复制胁迫和内源ROS过量的癌细胞带来益处(例如PARP1和MTH1的抑制剂)。如果抑制剂靶向抗凋亡蛋白和通路,可以想见基因毒剂和生物疗法致使细胞死亡的门限将降低。未来,对于特异DNA损伤如何激活和协调生与死的互戏还需要详细研究。在复制和转录时的修复和处置,以及它们如何激活求生寻死通路,对于癌症治疗异常重要。这些研究的终极目标是在用基因毒剂治疗癌症时保护正常组织而让癌细胞去死。保护正常组织对干细胞极其重要,因为干细胞更容易激活DNA损伤诱导的凋亡[185];保护正常组织对消除正常群体里的基因组损伤细胞也极为重要,而这也是一种防癌的策略。
致谢
作者之工作受某和某之资助。
利益纠纷陈述
作者宣称无利益纠纷。
参考文献
1. Voulgaridou, G. P.,Anestopoulos, I., Franco, R., Panayiotidis, M. I. & Pappa, A. DNA damageinduced by endogenous aldehydes: current state of knowledge. Mutat. Res. 711,13–27 (2011).
2. Fialkow, L., Wang, Y. & Downey, G. P.Reactive oxygen and nitrogen species as signaling molecules regulatingneutrophil function. Free Radic. Biol. Med. 42, 153–164 (2007).
3. Kielbassa, C., Roza, L. & Epe, B. Wavelengthdependence of oxidative DNA damage induced by UV and visible light.Carcinogenesis 18, 811–816 (1997).
4. Cadet, J., Ravanat, J. L., TavernaPorro, M.,Menoni, H. & Angelov, D. Oxidatively generated complex DNA damage: tandemand clustered lesions. Cancer Lett. 327, 5–15 (2012).
5. Roos, W. P. & Kaina, B. DNA damage-inducedcell death: from specific DNA lesions to the DNA damage response and apoptosis.Cancer Lett. 332, 237–248 (2013).
6. Dipple, A. DNA adducts of chemical carcinogens.Carcinogenesis 16, 437–441 (1995).
7. Miller, E. C. & Miller, J. A. Mechanisms ofchemical carcinogenesis. Cancer 47, 1055–1064 (1981).
8. Hoeijmakers, J. H. Genome maintenance mechanismsfor preventing cancer. Nature 411, 366–374 (2001).
9. Roos, W. P. & Kaina, B. DNA damage-inducedcell death by apoptosis. Trends Mol. Med. 12, 440–450 (2006).
10. Lowe, S. W. & Lin, A. W. Apoptosis incancer. Carcinogenesis 21, 485–495 (2000).
11. Fulda, S. Cell death and survival signaling inoncogenesis. Klin. Padiatr. 222, 340–344 (2010).
12. Helleday, T., Petermann, E., Lundin, C.,Hodgson, B. & Sharma, R. A. DNA repair pathways as targets for cancertherapy. Nat. Rev. Cancer 8, 193–204 (2008).
13. Fu, D., Calvo, J. A. & Samson, L. D.Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat.Rev. Cancer 12, 104–120 (2012).
14. Kaina, B., Christmann, M., Naumann, S. &Roos, W. P. MGMT: key node in the battle against genotoxicity, carcinogenicityand apoptosis induced by alkylating agents. DNA Repair (Amst.) 6, 1079–1099(2007).
15. Ensminger, M. et al. DNA breaks and chromosomalaberrations arise when replication meets base excision repair. J. Cell Biol.206, 29–43 (2014).
16. Zeman, M. K. & Cimprich, K. A. Causes andconsequences of replication stress. Nat. Cell Biol. 16, 2–9 (2014).
17. Halazonetis, T. D., Gorgoulis, V. G. &Bartek, J. An oncogene-induced DNA damage model for cancer development. Science319, 1352–1355 (2008).
18. Ando, K. et al. PIDD death-domainphosphorylation by ATM controls prodeath versus prosurvival PIDDosomesignaling. Mol. Cell 47, 681–693 (2012).
19. Lips, J. & Kaina, B. DNA double-strandbreaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis 22,579–585 (2001).
20. Virag, L., Robaszkiewicz, A., Rodriguez-Vargas,J. M. & Oliver, F. J. Poly(ADP-ribose) signaling in cell death. Mol.Aspects Med. 34, 1153–1167 (2013).
21. Chiu, L. Y., Ho, F. M., Shiah, S. G., Chang, Y.& Lin, W. W. Oxidative stress initiates DNA damager MNNG-inducedpoly(ADP-ribose)polymerase-1-dependent parthanatos cell death. Biochem.Pharmacol. 81, 459–470 (2011).
22. Zhou, Z. D., Chan, C. H., Xiao, Z. C. & Tan,E. K. Ring finger protein 146/Iduna is a poly(ADP-ribose) polymer binding andPARsylation dependent E3 ubiquitin ligase. Cell Adh. Migr. 5, 463–471 (2011).
23. Roos, W. P. et al. The translesion polymeraseRev3L in the tolerance of alkylating anticancer drugs. Mol. Pharmacol. 76,927–934 (2009).
24. Ashour, M. E., Atteya, R. & El-Khamisy, S.F. Topoisomerase-mediated chromosomal break repair: an emerging player in manygames. Nat. Rev. Cancer 15, 137–151 (2015).
25. Stingele, J., Habermann, B. & Jentsch, S.DNA–protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem.Sci. 40, 67–71 (2015).
26. Rogakou, E. P., Pilch, D. R., Orr, A. H.,Ivanova, V. S. & Bonner, W. M. DNA double-stranded breaks induce histoneH2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998).Identification of a specific marker of DSBs, namely, the phosphorylated form ofH2AX (γH2AX), greatly stimulated research on DNAdamage. γH2AX is currently the most sensitive markerfor DSBs and blocked replication forks.
27. Eich, M., Roos, W. P., Nikolova, T. & Kaina,B. Contribution of ATM and ATR to the resistance of glioblastoma and malignantmelanoma cells to the methylating anticancer drug temozolomide. Mol. CancerTher. 12, 2529–2540 (2013).
28. Stankovic, T. et al. ATM mutations in sporadiclymphoid tumours. Leuk. Lymphoma 43, 1563–1571 (2002).
29. Kim, H. et al. Having pancreatic cancer withtumoral loss of ATM and normal TP53 protein expression is associated with apoorer prognosis. Clin. Cancer Res. 20, 1865–1872 (2014).
30. Pusapati, R. V. et al. ATM promotes apoptosisand suppresses tumorigenesis in response to Myc. Proc. Natl Acad. Sci. USA 103,1446–1451 (2006).
31. Bitomsky, N. & Hofmann, T. G. Apoptosis andautophagy: regulation of apoptosis by DNA damage signalling — roles of p53, 73and HIPK2. FEBS J. 276, 6074–6083 (2009).
32. Dahal, G. R. et al. Caspase-2 cleaves DNAfragmentation factor (DFF45)/inhibitor of caspase-activated DNase (ICAD). Arch.Biochem. Biophys. 468, 134–139 (2007). Here, the authors demonstrated a directlink between nuclear caspase 2 and the apoptosis-triggering nuclease CAD.
33. Bernstein, C., Bernstein, H., Payne, C. M. &Garewal, H. DNA repair/pro-apoptotic dual-role proteins in five major DNArepair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 511,145–178 (2002).
34. Xu, Y. & Baltimore, D. Dual roles of ATM inthe cellular response to radiation and in cell growth control. Genes Dev. 10,2401–2410 (1996). In this paper it is shown, using Atm-knockout mice, that ATMplays a role in the DDR to ionizing radiation.
35. Swift, M., Morrell, D., Massey, R. B. &Chase, C. L. Incidence of cancer in 161 families affected byataxia-telangiectasia. N. Engl. J. Med. 325, 1831–1836 (1991).
36. Thompson, D. et al. Cancer risks and mortalityin heterozygous ATM mutation carriers. J. Natl Cancer Inst. 97, 813–822 (2005).
37. Tanaka, A. et al. Germline mutation in ATR inautosomal- dominant oropharyngeal cancer syndrome. Am. J. Hum. Genet. 90,511–517 (2012). In references 35–37, evidence is provided that mutations in ATMand ATR can predispose to cancer development.
38. Khanna, K. K. Cancer risk and the ATM gene: acontinuing debate. J. Natl Cancer Inst. 92, 795–802 (2000).
39. Galluzzi, L., Vitale, I., Vacchelli, E. &Kroemer, G. Cell death signaling and anticancer therapy. Front. Oncol. 1, 5(2011).
40. Helleday, T. Homologous recombination in cancerdevelopment, treatment and development of drug resistance. Carcinogenesis 31,955–960 (2010).
41. Sale, J. E. Competition, collaboration andcoordination—determining how cells bypass DNA damage. J. Cell Sci. 125,1633–1643 (2012).
42. Vandenabeele, P., Galluzzi, L., Vanden Berghe,T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellularexplosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010).
43. Bartek, J. & Lukas, J. DNA damagecheckpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol.19, 238–245 (2007).
44. Vakifahmetoglu, H., Olsson, M. &Zhivotovsky, B. Death through a tragedy: mitotic catastrophe. Cell DeathDiffer. 15, 1153–1162 (2008).
45. Lahav, G. et al. Dynamics of the p53-Mdm2feedback loop in individual cells. Nat. Genet. 36, 147–150 (2004).
46. Zhang, X. P., Liu, F., Cheng, Z. & Wang, W.Cell fate decision mediated by p53 pulses. Proc. Natl Acad. Sci. USA 106,12245–12250 (2009).
47. Tian, X. J., Liu, F., Zhang, X. P., Li, J. &Wang, W. A two-step mechanism for cell fate decision by coordination of nuclearand mitochondrial p53 activities. PLoS ONE 7, e38164 (2012).
48. Inga, A., Storici, F., Darden, T. A. &Resnick, M. A. Differential transactivation by the p53 transcription factor ishighly dependent on p53 level and promoter target sequence. Mol. Cell. Biol.22, 8612–8625 (2002). The differential binding affinity of p53 for targetpromoters and its contribution to the different functions of p53 wasdemonstrated in this work.
49. Nicol, S. M. et al. The RNA helicase p68 (DDX5)is selectively required for the induction of p53-dependent p21 expression andcell-cycle arrest after DNA damage. Oncogene 32, 3461–3469 (2013).
50. Tanaka, T. Ohkubo, S., Tatsuno, I. & Prives,C. hCAS/CSE1L associates with chromatin and regulates expression of select p53target genes. Cell 130, 638–650 (2007).
51. Sullivan, A. & Lu, X. ASPP: a new family ofoncogenes and tumour suppressor genes. Br. J. Cancer 96, 196–200 (2007).
52. Lettre, G. et al. Genome-wide RNAi identifiesp53-dependent and -independent regulators of germ cell apoptosis in C. elegans.Cell Death Differ. 11, 1198–1203 (2004).
53. Loughery, J., Cox, M., Smith, L. M. & Meek,D. W. Critical role for p53-serine 15 phosphorylation in stimulatingtransactivation at p53-responsive promoters. Nucleic Acids Res. 42, 7666–7680(2014).
54. Jabbur, J. R., Huang, P. & Zhang, W. DNAdamage-induced phosphorylation of p53 at serine 20 correlates with p21 andMdm-2 induction in vivo. Oncogene 19, 6203–6208 (2000). References 53 and 54show that p53 phosphorylated at Ser15 and Ser20 displays different roles inauto-regulation and cell cycle checkpoint activation.
55. Pietsch, E. C., Sykes, S. M., McMahon, S. B.& Murphy, M. E. The p53 family and programmed cell death. Oncogene 27,6507–6521 (2008).
56. Ichwan, S. J. et al. Defect in serine 46phosphorylation of p53 contributes to acquisition of p53 resistance in oralsquamous cell carcinoma cells. Oncogene 25, 1216–1224 (2006).
57. Mayo, L. D. et al. Phosphorylation of human p53at serine 46 determines promoter selection and whether apoptosis is attenuatedor amplified. J. Biol. Chem. 280, 25953–25959 (2005). In this paper it is shownthat p53 phosphorylated at Ser46 specifically activates pro-apoptotic genes.
58. Oda, K. et al. p53AIP1, a potential mediator ofp53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell102, 849–862 (2000).
59. Hofmann, T. G. et al. Regulation of p53 activityby its interaction with homeodomain-interacting protein kinase-2. Nat. CellBiol. 4, 1–10 (2002). In this paper, it is shown for the first time that HIPK2is responsible for phosphorylating p53 at Ser46 in response to DNA damage.
60. Bulavin, D. V. et al. Phosphorylation of humanp53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis inresponse to UV radiation. EMBO J. 18, 6845–6854 (1999).
61. Yoshida, K., Liu, H. & Miki, Y. Proteinkinase Cδ regulates Ser46 phosphorylation of p53 tumorsuppressor in the apoptotic response to DNA damage. J. Biol. Chem. 281, 5734–5740 (2006).
62. Okamura, S. et al. p53DINP1, a p53-induciblegene, regulates p53-dependent apoptosis. Mol. Cell 8, 85–94 (2001).
63. Taira, N., Nihira, K., Yamaguchi, T., Miki, Y.& Yoshida, K. DYRK2 is targeted to the nucleus and controls p53 via Ser46phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725–738(2007).
64. Lee, M. G. et al. XAF1 directs apoptotic switchof p53 signaling through activation of HIPK2 and ZNF313. Proc. Natl Acad. Sci.USA 111, 15532–15537 (2014).
65. Winter, M. et al. Control of HIPK2 stability byubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat. Cell Biol. 10,812–824 (2008). This paper shows that HIPK2 is part of the DDR.
66. Guo, A. et al. The function of PML inp53-dependent apoptosis. Nat. Cell Biol. 2, 730–736 (2000).
67. Takekawa, M. et al. p53-inducible Wip1phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signalingin response to UV radiation. EMBO J. 19, 6517–6526 (2000).
68. Nakayama, K., Qi, J. & Ronai, Z. Theubiquitin ligase Siah2 and the hypoxia response. Mol. Cancer Res. 7, 443–451(2009).
69. Zhao, S. et al.Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity andinduce HIF-1α. Science 324, 261–265(2009).
70. Bulavin, D. V. et al. Amplification of PPM1D inhuman tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 31, 210–215(2002).
71. Yip, K. W. & Reed, J. C. Bcl-2 familyproteins and cancer. Oncogene 27, 6398–6406 (2008).
72. Tamm, I. et al. IAP-family protein survivininhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases,and anticancer drugs. Cancer Res. 58, 5315–5320 (1998).
73. Ambrosini, G., Adida, C. & Altieri, D. C. Anovel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat.Med. 3, 917–921 (1997). Here, it was demonstrated that survivin, which wasthought to only have a role during fetal development, is overexpressed intumours, having a role in apoptosis regulation.
74. Knauer, S. K., Mahendrarajah, N., Roos, W. P.& Kramer, O. H. The inducible E3 ubiquitin ligases SIAH1 and SIAH2 performcritical roles in breast and prostate cancers. Cytokine Growth Factor Rev. 26,405–413 (2015).
75. Andera, L. & Wasylyk, B. Transcriptionabnormalities potentiate apoptosis of normal human fibroblasts. Mol. Med. 3,852–863 (1997).
76. De Carvalho, D. D. et al. DNA methylationscreening identifies driver epigenetic events of cancer cell survival. CancerCell 21, 655–667 (2012).
77. Ljungman, M. & Lane, D. P. Transcription —guarding the genome by sensing DNA damage. Nat. Rev. Cancer 4, 727–737 (2004).
78. Derheimer, F. A. et al. RPA and ATR linktranscriptional stress to p53. Proc. Natl Acad. Sci. USA 104, 12778–12783(2007). This paper showed convincingly that DNA lesions blocking transcriptionactivate ATR and consequently the DDR.
79. Conforti, G., Nardo, T., D'Incalci, M. &Stefanini, M. Proneness to UV-induced apoptosis in human fibroblasts defectivein transcription coupled repair is associated with the lack of Mdm2transactivation. Oncogene 19, 2714–2720 (2000).
80. Hwang, B. J., Ford, J. M., Hanawalt, P. C. &Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent andis involved in global genomic repair. Proc. Natl Acad. Sci. USA 96, 424–428(1999).
81. Barckhausen, C., Roos, W. P., Naumann, S. C.& Kaina, B. Malignant melanoma cells acquire resistance to DNA interstrandcross-linking chemotherapeutics by p53-triggered upregulation ofDDB2/XPC-mediated DNA repair. Oncogene 33, 1964–1974 (2014).
82. Batista, L. F., Roos, W. P., Christmann, M.,Menck, C. F. & Kaina, B. Differential sensitivity of malignant glioma cellsto methylating and chloroethylating anticancer drugs: 53 determines the switchby regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 67,11886–11895 (2007). Here, the dependence on the anticancer drug to activate thedual function of p53 in regulating apoptosis or DNA repair is demonstrated.
83. Proietti De Santis, L. et al. Transcriptioncoupled repair efficiency determines the cell cycle progression and apoptosisafter UV exposure in hamster cells. DNA Repair (Amst.) 1, 209–223 (2002).
84. Ljungman, M., O'Hagan, H. M. & Paulsen, M.T. Induction of ser15 and lys382 modifications of p53 by blockage oftranscription elongation. Oncogene 20, 5964–5971 (2001).
85. Herrlich, P. et al. The mammalian UV response:mechanism of DNA damage induced gene expression. Adv. Enzyme Regul. 34, 381–395(1994).
86. Tomicic, M. T. et al. Delayed c-Fos activationin human cells triggers XPF induction and an adaptive response to UVC-inducedDNA damage and cytotoxicity. Cell. Mol. Life Sci. 68, 1785–1798 (2011).
87. Haas, S. & Kaina, B. c-Fos is involved inthe cellular defence against the genotoxic effect of UV radiation. Carcinogenesis16, 985–991 (1995).
88. Kaina, B., Haas, S. & Kappes, H. A generalrole for c-Fos in cellular protection against DNA-damaging carcinogens andcytostatic drugs. Cancer Res. 57, 2721–2731 (1997).
89. Christmann, M., Tomicic, M. T., Origer, J.,Aasland, D. & Kaina, B. c-Fos is required for excision repair of UV-lightinduced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Res.34, 6530–6539 (2006). In references 86–89, evidence is provided that FOS has adual role in regulating DNA repair and, if repair is saturated, apoptosis.
90. Christmann, M. & Kaina, B. Transcriptionalregulation of human DNA repair genes following genotoxic stress: triggermechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res.41, 8403–8420 (2013).
91. Marteijn, J. A., Lans, H., Vermeulen, W. &Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles incancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481 (2014).
92. Hamdi, M. et al. DNA damage in transcribed genesinduces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene24, 7135–7144 (2005). The fundamental relationship between NER, JNK, MKP1 andapoptosis was elucidated in this article.
93. Brozovic, A. et al. Long-term activation ofSAPK/JNK, 38 kinase and fas-L expression by cisplatin is attenuated in humancarcinoma cells that acquired drug resistance. Int. J. Cancer 112, 974–985(2004).
94. Hirsch, D. D. & Stork, P. J.Mitogen-activated protein kinase phosphatases inactivate stress-activatedprotein kinase pathways in vivo. J. Biol. Chem. 272, 4568–4575 (1997).
95. Christmann, M., Tomicic, M. T., Aasland, D.& Kaina, B. A role for UV-light-induced c-Fos: stimulation of nucleotideexcision repair and protection against sustained JNK activation and apoptosis.Carcinogenesis 28, 183–190 (2007).
96. Roos, W. P. et al. Intrinsic anticancer drugresistance of malignant melanoma cells is abrogated by IFN-βand valproic acid. Cancer Res. 71, 4150–4160 (2011).
97. Christmann, M., Verbeek, B., Roos, W. P. &Kaina, B. O6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues andtumors: enzyme activity, promoter methylation and immunohistochemistry.Biochim. Biophys. Acta 1816, 179–190 (2011).
98. Weller, M. et al. MGMT promoter methylation inmalignant gliomas: ready for personalized medicine? Nat. Rev. Neurol. 6, 39–51(2010).
99. Smith, J. Human Sir2 and the 'silencing' of p53activity. Trends Cell Biol. 12, 404–406 (2002).
100. Chen, W. Y. et al. Tumor suppressor HIC1directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell123, 437–448 (2005). In this paper, the feedback loop between p53, HIC1 and thedeacetylase SIRT1 was described.
101. Soengas, M. S. et al. Inactivation of theapoptosis effector Apaf-1 in malignant melanoma. Nature 409, 207–211 (2001).
102. von Zglinicki, T., Saretzki, G., Ladhoff, J.,d'Adda di Fagagna, F. & Jackson, S. P. Human cell senescence as a DNAdamage response. Mech. Ageing Dev. 126, 111–117 (2005).
103. Stevenson, A. F. & Cremer, T. Senescence invitro and ionising radiations—the human diploid fibroblast model. Mech. AgeingDev. 15, 51–63 (1981).
104. Rodier, F. et al. Persistent DNA damagesignalling triggers senescence-associated inflammatory cytokine secretion. Nat.Cell Biol. 11, 973–979 (2009).
105. Mirzayans, R., Andrais, B., Hansen, G. &Murray, D. Role of p16INK4A in replicative senescence and DNA damage-inducedpremature senescence in p53-deficient human cells. Biochem. Res. Int. 2012,951574 (2012).
106. Broccoli, D., Smogorzewska, A., Chong, L. &de Lange, T. Human telomeres contain two distinct Myb-related proteins, TRF1 andTRF2. Nat. Genet. 17, 231–235 (1997).
107. Fumagalli, M. et al. Telomeric DNA damage isirreparable and causes persistent DNA-damage-response activation. Nat. CellBiol. 14, 355–365 (2012). The authors demonstrated that the shelterin proteinTRF2 prevents the completion of DSB repair by NHEJ in telomeres by inhibitingligase IV, thereby causing persistent ATM-mediated DDR signalling, leading tosenescence.
108. van Steensel, B., Smogorzewska, A. & deLange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 92,401–413 (1998).
109. Brunori, M. et al. TRF2 inhibition promotesanchorage-independent growth of telomerase-positive human fibroblasts. Oncogene25, 990–997 (2006).
110. Hundley, J. E. et al. Increased tumor proliferationand genomic instability without decreased apoptosis in MMTV-ras mice deficientin p53. Mol. Cell. Biol. 17, 723–731 (1997).
111. Knizhnik, A. V. et al. Survival and deathstrategies in glioma cells: autophagy, senescence and apoptosis triggered by asingle type of temozolomide-induced DNA damage. PLoS ONE 8, e55665 (2013).
112. Stambolic, V. et al. Regulation of PTENtranscription by p53. Mol. Cell 8, 317–325 (2001).
113. Crighton, D. et al. DRAM, a p53-inducedmodulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006).
114. Ravikumar, B. et al. Raised intracellularglucose concentrations reduce aggregation and cell death caused by mutantHuntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy.Hum. Mol. Genet. 12, 985–994 (2003).
115. Cao, C. et al. Inhibition of mammalian target ofrapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN nullprostate cancer cells. Cancer Res. 66, 10040–10047 (2006).
116. Mah, L. Y., O'Prey, J., Baudot, A. D., Hoekstra,A. & Ryan, K. M. DRAM-1 encodes multiple isoforms that regulate autophagy.Autophagy 8, 18–28 (2012).
117. de Murcia, J. M. et al. Requirement ofpoly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells.Proc. Natl Acad. Sci. USA 94, 7303–7307 (1997).
118. Olson, R. D., Boerth, R. C., Gerber, J. G. &Nies, A. S. Mechanism of adriamycin cardiotoxicity: evidence for oxidativestress. Life Sci. 29, 1393–1401 (1981).
119. Munoz-Gamez, J. A. et al. PARP-1 is involved inautophagy induced by DNA damage. Autophagy 5, 61–74 (2009).
120. Rodriguez-Vargas, J. M. et al. ROS-induced DNAdamage and PARP-1 are required for optimal induction of starvation-inducedautophagy. Cell Res. 22, 1181–1198 (2012). References 117, 119 and 120 describethe fundamental role of PARP1 for survival and regulation of autophagyfollowing DNA damage.
121. Li, L., Ishdorj, G. & Gibson, S. B. Reactiveoxygen species regulation of autophagy in cancer: implications for cancertreatment. Free Radic. Biol. Med. 53, 1499–1410 (2012).
122. Lamore, S. D. & Wondrak, G. T.Autophagic-lysosomal dysregulation downstream of cathepsin B inactivation inhuman skin fibroblasts exposed to UVA. Photochem. Photobiol. Sci. 11, 163–172(2012).
123. Suzuki, A. et al. IGF-1 phosphorylates AMPK-α subunit in ATM-dependent and LKB1-independent manner.Biochem. Biophys. Res. Commun. 324, 986–992 (2004).
124. Ochs, K. & Kaina, B. Apoptosis induced byDNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated andFas/caspase-8 independent. Cancer Res. 60, 5815–5824 (2000).
125. Caporali, S. et al. DNA damage induced bytemozolomide signals to both ATM and ATR: role of the mismatch repair system.Mol. Pharmacol. 66, 478–491 (2004).
126. Alexander, A., Kim, J. & Walker, C. L. ATMengages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 6,672–673 (2010).
127. Alexander, A. et al. ATM signals to TSC2 in thecytoplasm to regulate mTORC1 in response to ROS. Proc. Natl Acad. Sci. USA 107,4153–4158 (2010). This paper describes the link between ATM and mTOR.
128. Park, C., Suh, Y. & Cuervo, A. M. Regulateddegradation of Chk1 by chaperone-mediated autophagy in response to DNA damage.Nat. Commun. 6, 6823 (2015).
129. Pattingre, S. et al. Bcl-2 antiapoptoticproteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 (2005).
130. Wei, Y., Sinha, S. & Levine, B. Dual role ofJNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation.Autophagy 4, 949–951 (2008).
131. Tsujimoto, Y. & Shimizu, S. Another way todie: autophagic programmed cell death. Cell Death Differ. 12 (Suppl. 2),1528–1534 (2005).
132. Tsai, W. B., Chung, Y. M., Takahashi, Y., Xu, Z.& Hu, M. C. Functional interaction between FOXO3a and ATM regulates DNAdamage response. Nat. Cell Biol. 10, 460–467 (2008).
133. Kanzawa, T., Kondo, Y., Ito, H., Kondo, S. &Germano, I. Induction of autophagic cell death in malignant glioma cells byarsenic trioxide. Cancer Res. 63, 2103–2108 (2003).
134. Isakson, P., Bjoras, M., Boe, S. O. &Simonsen, A. Autophagy contributes to therapy-induced degradation of thePML/RARA oncoprotein. Blood 116, 2324–2331 (2010).
135. Rez, G., Toth, S. & Palfia, Z. Cellularautophagic capacity is highly increased in azaserine-induced premalignantatypical acinar nodule cells. Carcinogenesis 20, 1893–1898 (1999).
136. Yang, P. M. & Chen, C. C. Life or death?Autophagy in anticancer therapies with statins and histone deacetylaseinhibitors. Autophagy 7, 107–108 (2011).
137. Gozuacik, D. & Kimchi, A. Autophagy as acell death and tumor suppressor mechanism. Oncogene 23, 2891–2906 (2004).
138. Roos, W. P. et al. Apoptosis in malignant gliomacells triggered by the temozolomide-induced DNA lesion O6-methylguanine.Oncogene 26, 186–197 (2007).
139. Altieri, D. C. Survivin and IAP proteins incell-death mechanisms. Biochem. J. 430, 199–205 (2010).
140. Adida, C. et al. Developmentally regulatedexpression of the novel cancer anti-apoptosis gene survivin in human and mousedifferentiation. Am. J. Pathol. 152, 43–49 (1998).
141. Santa Cruz Guindalini, R., Mathias Machado, M.C. & Garicochea, B. Monitoring survivin expression in cancer: implicationsfor prognosis and therapy. Mol. Diagn. Ther. 17, 331–342 (2013).
142. Greve, B. et al. Survivin, a target to modulatethe radiosensitivity of Ewing's sarcoma. Strahlenther. Onkol. 188, 1038–1047(2012).
143. Wallace, M. D., Southard, T. L., Schimenti, K.J. & Schimenti, J. C. Role of DNA damage response pathways in preventingcarcinogenesis caused by intrinsic replication stress. Oncogene 33, 3688–3695(2014).
144. Wu, Y. K. et al. Nuclear survivin expression: aprognostic factor for the response to taxane-platinum chemotherapy in patientswith advanced non-small cell lung cancer. Med. Oncol. 31, 79 (2014).
145. Sedlak, T. W. et al. Multiple Bcl-2 familymembers demonstrate selective dimerizations with Bax. Proc. Natl Acad. Sci. USA92, 7834–7838 (1995).
146. Boehme, K. A., Kulikov, R. & Blattner, C.p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc.Natl Acad. Sci. USA 105, 7785–7790 (2008).
147. Li, Y., Xiong, H. & Yang, D. Q. Functionalswitching of ATM: sensor of DNA damage in proliferating cells and mediator ofAkt survival signal in post-mitotic human neuron-like cells. Chin. J. Cancer31, 364–372 (2012).
148. Caporali, S. et al. AKT is activated in anataxia-telangiectasia and Rad3-related-dependent manner in response totemozolomide and confers protection against drug-induced cell growthinhibition. Mol. Pharmacol. 74, 173–183 (2008).
149. Fraser, M. et al. MRE11 promotes AKTphosphorylation in direct response to DNA double-strand breaks. Cell Cycle 10,2218–2232 (2011).
150. Datta, S. R. et al. Akt phosphorylation of BADcouples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241(1997).
151. Kim, A. H., Khursigara, G., Sun, X., Franke, T.F. & Chao, M. V. Akt phosphorylates and negatively regulates apoptosissignal-regulating kinase 1. Mol. Cell. Biol. 21, 893–901 (2001).
152. Cardone, M. H. et al. Regulation of cell deathprotease caspase-9 by phosphorylation. Science 282, 1318–1321 (1998).
153. Mayo, L. D. & Donner, D. B. Aphosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 fromthe cytoplasm to the nucleus. Proc. Natl Acad. Sci. USA 98, 11598–11603 (2001).
154. Wirawan, E. et al. Caspase-mediated cleavage ofBeclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis bypromoting the release of proapoptotic factors from mitochondria. Cell DeathDis. 1, e18 (2010). The importance of caspase cleavage of Beclin 1 in theswitch between autophagy and apoptosis is demonstrated in this paper.
155. Trichonas, G. et al. Receptor interactingprotein kinases mediate retinal detachment-induced photoreceptor necrosis and compensatefor inhibition of apoptosis. Proc. Natl Acad. Sci. USA 107, 21695–21700 (2010).
156. Osborn, S. L. et al. Fas-associated death domain(FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc.Natl Acad. Sci. USA 107, 13034–13039 (2010).
157. Vanlangenakker, N., Bertrand, M. J., Bogaert,P., Vandenabeele, P. & Vanden Berghe, T. TNF-induced necroptosis in L929cells is tightly regulated by multiple TNFR1 complex I and II members. CellDeath Dis. 2, e230 (2011).
158. Chan, F. K. et al. A role for tumor necrosisfactor receptor-2 and receptor-interacting protein in programmed necrosis andantiviral responses. J. Biol. Chem. 278, 51613–51621 (2003).
159. Hu, X., Han, W. & Li, L. Targeting the weakpoint of cancer by induction of necroptosis. Autophagy 3, 490–492 (2007).
160. Skulachev, V. P. Bioenergetic aspects ofapoptosis, necrosis and mitoptosis. Apoptosis 11, 473–485 (2006).
161. Los, M. et al. Activation and caspase-mediatedinhibition of PARP: a molecular switch between fibroblast necrosis andapoptosis in death receptor signaling. Mol. Biol. Cell 13, 978–988 (2002).
162. Newton, K. et al. Activity of protein kinaseRIPK3 determines whether cells die by necroptosis or apoptosis. Science 343,1357–1360 (2014).
163. Duprez, L. et al. Intermediate domain ofreceptor-interacting protein kinase 1 (RIPK1) determines switch betweennecroptosis and RIPK1 kinase-dependent apoptosis. J. Biol. Chem. 287,14863–14872 (2012). In references 161–163, the role of PARP1 and RIPK3–RIPK1was demonstrated in the switch between necroptosis and apoptosis.
164. Upton, J. W., Kaiser, W. J. & Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmednecrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11,290–297 (2012).
165. Kaiser, W. J. & Offermann, M. K. Apoptosisinduced by the toll-like receptor adaptor TRIF is dependent on its receptorinteracting protein homotypic interaction motif. J. Immunol. 174, 4942–4952(2005).
166. O'Donnell, M. A. et al. Caspase 8 inhibitsprogrammed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 (2011).
167. He, M. X. & He, Y. W. A role for c-FLIP(L)in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes.Cell Death Differ. 20, 188–197 (2013).
168. McComb, S. et al. cIAP1 and cIAP2 limitmacrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell DeathDiffer. 19, 1791–1801 (2012).
169. Cai, Z. et al. Plasma membrane translocation oftrimerized MLKL protein is required for TNF-induced necroptosis. Nat. CellBiol. 16, 55–65 (2014).
170. Andrabi, S. A., Dawson, T. M. & Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. NYAcad. Sci. 1147, 233–241 (2008).
171. Burkle, A. Poly(ADP-ribose). The most elaboratemetabolite of NAD+. FEBS J. 272, 4576–4589 (2005).
172. Szabo, C. & Dawson, V. L. Role ofpoly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. TrendsPharmacol. Sci. 19, 287–298 (1998).
173. Ying, W., Garnier, P. & Swanson, R. A. NAD+repletion prevents PARP-1-induced glycolytic blockade and cell death incultured mouse astrocytes. Biochem. Biophys. Res. Commun. 308, 809–813 (2003).
174. Osato, K. et al. Apoptosis-inducing factordeficiency decreases the proliferation rate and protects the subventricularzone against ionizing radiation. Cell Death Dis. 1, e84 (2010).
175. Leist, M., Single, B., Castoldi, A. F., Kuhnle,S. & Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration:a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185,1481–1486 (1997). This paper shows for the first time that the ATP level determinesthe switch between apoptosis and necrosis.
176. Huang, C. T., Huang, D. Y., Hu, C. J., Wu, D.& Lin, W. W. Energy adaptive response during parthanatos is enhanced byPD98059 and involves mitochondrial function but not autophagy induction.Biochim. Biophys. Acta 1843, 531–543 (2013).
177. Kerr, J. F., Wyllie, A. H. & Currie, A. R.Apoptosis: a basic biological phenomenon with wide-ranging implications intissue kinetics. Br. J. Cancer 26, 239–257 (1972). These authors described forthe first time that apoptosis is an important cell death mechanism.
178. Kroemer, G. et al. Classification of cell death:recommendations of the Nomenclature Committee on Cell Death 2009. Cell DeathDiffer. 16, 3–11 (2009).
179. Tang, H. L., Yuen, K. L., Tang, H. M. &Fung, M. C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 100,118–122 (2009).
180. Maynard, S. et al. Human embryonic stem cellshave enhanced repair of multiple forms of DNA damage. Stem Cells 26, 2266–2274(2008).
181. Bauer, M. et al. Human monocytes are severelyimpaired in base and DNA double-strand break repair that renders themvulnerable to oxidative stress. Proc. Natl Acad. Sci. USA 108, 21105–21110 (2011).
182. Narciso, L. et al. Terminally differentiatedmuscle cells are defective in base excision DNA repair and hypersensitive tooxygen injury. Proc. Natl Acad. Sci. USA 104, 17010–17015 (2007).
183. Kauffmann, A. et al. High expression of DNArepair pathways is associated with metastasis in melanoma patients. Oncogene27, 565–573 (2008).
184. Tomicic, M. T. et al. Translesion polymerase etais upregulated by cancer therapeutics and confers anticancer drug resistance.Cancer Res. 74, 5585–5596 (2014).
185. Roos, W. P., Christmann, M., Fraser, S. T. &Kaina, B. Mouse embryonic stem cells are hypersensitive to apoptosis triggeredby the DNA damage O6-methylguanine due to high E2F1 regulated mismatch repair.Cell Death Differ. 14, 1422–1432 (2007).
186. Volcic, M. et al. NF-κBregulates DNA double-strand break repair in conjunction with BRCA1-CtIPcomplexes. Nucleic Acids Res. 40, 181–195 (2012).
187. Huang, T. T., Wuerzberger-Davis, S. M., Wu, Z.H. & Miyamoto, S. Sequential modification of NEMO/IKKγby SUMO-1 and ubiquitin mediates NF-κB activation bygenotoxic stress. Cell 115, 565–576 (2003).
188. Cai, Q. & Robertson, E. S. Ubiquitin/SUMOmodification regulates VHL protein stability and nucleocytoplasmiclocalization. PLoS ONE 5, e12636 (2010).
189. Hur, G. M. et al. The death domain kinase RIPhas an essential role in DNA damage-induced NF-κBactivation. Genes Dev. 17, 873–882 (2003).
190. Brzoska, K. & Szumiel, I. Signalling loops and linear pathways: NF-κB activation in response to genotoxic stress. Mutagenesis24, 1–8 (2009).
191. Lee, H. H., Dadgostar, H., Cheng, Q., Shu, J.& Cheng, G. NF-κB-mediated up-regulation of Bcl-xand Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc.Natl Acad. Sci. USA 96, 9136–9141 (1999).
192. Chu, Z. L. et al. Suppression of tumor necrosisfactor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaBcontrol. Proc. Natl Acad. Sci. USA 94, 10057–10062 (1997).
193. Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel,D. V. & Baldwin, A. S. Jr. NF-kappaB antiapoptosis: induction of TRAF1 andTRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281,1680–1683 (1998).
194. Zhou, A., Scoggin, S., Gaynor, R. B. &Williams, N. S. Identification of NF-κB-regulated genesinduced by TNFα utilizing expression profiling and RNAinterference. Oncogene 22, 2054–2064 (2003).
195. Cusack, J. C. Jr et al. Enhancedchemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications forsystemic nuclear factor-κB inhibition. Cancer Res. 61,3535–3540 (2001).
196. Yao, R. & Cooper, G. M. Requirement forphosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growthfactor. Science 267, 2003–2006 (1995).
197. Wendel, H. G. et al. Survival signalling by Aktand eIF4E in oncogenesis and cancer therapy. Nature 428, 332–337 (2004).
198. Toulany, M. et al. Akt promotes post-irradiationsurvival of human tumor cells through initiation, progression, and terminationof DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 10,945–957 (2012).
附表:靶向DNA损伤后求生通路之关键酶的临床策略之简表,以及它们的临床效果和临床评估的阶段。
求生通路 | 药物 | 作用模式 | 功效举例 | 临床评估 |
DNA损伤响应 | ||||
ATM | KU-60019 | ATM竞争性抑制剂 | 辐射敏感化神经胶质瘤细胞[1] | 临床前期 |
ATR | VE-821 | TAR竞争性抑制剂 | 细胞敏感于拓扑异构酶所致之凋亡[2] | 临床前期 |
Chk1/Chk2 | UCN-01 | 抑制磷酸激酶活性 | 增加吉西他滨( gemcitabine )所致的结肠癌细胞凋亡[3] | 与carboplatin联用于晚期实体瘤的一期临床试验 |
细胞周期阻滞效应子 | ||||
Wee1 | MK-1775 | Wee1竞争性抑制剂 | 减少非小细胞肺癌移植瘤( xenografts )之生长[4] | 联合辐射或temozolomide治疗新诊断的或复发的多形性胶质母细胞瘤,一期临床试验 |
Cdc25 | IRC-083864 | 可能为Cdc25竞争性抑制剂 | 减少胰腺癌移植瘤之生长[5] | 临床前期 |
DNA修复 | ||||
MGMT | O6- benzylguanine, Lomeguatrib | MGMT诱饵底物 | 作为诱饵底物,消除MGMT修复能力[6] | 用于Iomeguatrib和temozolomide治疗结直肠癌的二期临床试验[7];用于temozolomide加irinotecan加O6-benzylguanine对成人复发性恶性胶质瘤的一期临床试验[8] |
HR | RI-1 | RAD51抑制剂 | 石细胞敏感于MMC[9] | 临床前期 |
HR | Mirin | 阻止MRN依赖的ATM活化 | 特异抑制Mre11[10] | 临床前期 |
PARP | Olaparib | 将PARP锁定于DNA | 增加肺癌移植瘤之放射物敏感性[11] | 联合Cediranib Maleate治疗复发性卵巢、输卵管、或腹膜癌或复发性三重阴性乳腺癌的一期二期临床试验 |
NHEJ | NU7026 | DNA-PK竞争性抑制剂 | 增加白血病细胞中拓扑异构酶II毒性的杀伤力[12] | 临床前期 |
MTH1 | TH588 | 结合MTH1活化位点将之抑制 | 利用癌细胞氧化还原调控的功能障碍[13] | 临床前期 |
凋亡启动 | ||||
XIAP | Embelin | 阻断其余Caspase-9和Smac的结合 | 在急性T细胞白血病细胞中增益etoposide所致细胞杀伤力[14] | 临床前期 |
NF-κB | Bortezomib | 蛋白酶体抑制剂,阻止IκBα的降解 | 当与irinotecan(Top I抑制剂)联用时,与对照组相比,肿瘤生长降低94%[15] | 治疗多发性骨髓瘤的二期联创实验 |
MKP-1 | NSC 95397 | 磷酸酶抑制剂 | 在乳腺腺癌细胞中增进paclitaxel所致之凋亡[16] | 临床前期 |
c-FLIP | OH14 (Licenced to Taziana Life Sciences) | 抑制c-Flip与FADD的抑制性结合 | 目前不可知 | 临床前期 |
Bcl-2 | ABT-199 | BH3模拟物 | 增进doxorubicin等化疗制剂对T细胞急性成淋巴细胞白血病细胞系的杀伤力[17] | 联合rituximab或bendamustine加MabThera/Riruxan治疗复发或耐药慢性淋巴细胞白血病的三期临床 |
Survivin | YM155 | 抑制Survivin表达 | 导致SK-NEP-1迁移性肾癌细胞的凋亡[18] | 联合docetaxel作为Her2阴性迁移性乳腺癌的一线疗法的二期临床试验 |
Akt | MK2206 | 抑制T308和S473的自我磷酸化 | 与抗肿瘤药物和受体酪氨酸抑制剂连用增进凋亡[19] | 联合dinaciclib用于不可手术之胰腺癌的一期临床试验 |
大限 | ||||
葡萄糖代谢 | 2-Deoxy-D- glucose | 抑制糖酵解 | 鼠类淋巴瘤模型中增进大限疗法的细胞杀伤力[20] | 临床前期 |
自噬 | ||||
一般性抑制剂 | Hydroxychloroquine | 溶酶体抑制剂 | 增加MCF7-RR和LCC9乳腺癌细胞对抗雌激素疗法的敏感性[21] | 乳腺癌治疗之二期临床试验 |
一般性抑制剂 | Chloroquine | 溶酶体抑制剂 | 增进5-fluorouracil对胆囊癌细胞的杀伤力[22] | 小细胞肺癌的一期二期临床试验的第四阶段 |
mTOR | Rapamycin | 抑制mTOR激活自噬 | 减少90%的NNK所致鼠类肺瘤的肿瘤多样性(tumor multiplicity)[23] | 联合corticosteroid用于复发性急性淋巴母细胞白血病的临床试验 |
Bcl-2 | ABT-199 | BH3模拟物 | 在治疗套细胞淋巴瘤细胞(mantle cell lymphoma cells)时,增加细胞对核苷类似物Acadesine的响应[24] | 联合rituximab或bendamustine加MabThera/Riruxan治疗复发或耐药慢性淋巴细胞白血病的三期临床 |
https://m.sciencenet.cn/blog-331314-1133598.html
上一篇:内部翻译资料:泛素与苏木在DNA损伤响应中发挥的调控作用
下一篇:实验技术:卵母细胞、COC和植入前胚胎的免疫荧光标记