博文
edgeR:处理数字基因表达数据的一个例子
|||
edgeR:处理数字基因表达数据的一个例子
作者:一枝梅
中国科学院成都成都生物研究所
编辑:熊荣川
六盘水师范学院生物信息学实验室
http://blog.sciencenet.cn/u/Bearjazz
edgeR是bioconductor中专门用于处理RNA-seq数据的基于R语言的软件包。
关于edgeR的更多细节,请猛击:
http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
写在前面的话:
AS大部分的生物信息分析工具,edgeR的操作非常简单;真正复杂的是其背后的生物学意义,在类似的分析当中,主要指的就是实验的设计。如果实验设计不合理,得到的结果就没有意义。此外,实验的重复也很重要。
本例的实验设计:
本例是一个2X2的方案,同一种组织来自两个不同样本,在两个treatments之下得到的基因表达数据。
准备工作:
Reads的过滤、mapping和计数。为了使结果最可靠,应该去掉非一一对应的reads。Mapping的区域首选为外显子(推荐用HTSeq-count来进行计数)。
数据读取:
1)读取数据的方法不局限,只需要将计数过得reads数据以矩阵形式读入即可。例如使用read.delim()来读取
>rawdata<-read.delim(‘count.txt’,check.name=FALSE,stringAsFactors=FALSE)
> head(rawdata)#检查输入是否正确
 FIG.1
FIG.1
>y<-DGEList(counts=rawdata[,2:5]),genes=rawdata[,1])
2)过滤与标准化
>keep<-rowSums(cpm(y)>1)>=4 #过滤标准为至少one count per million (cpm)
>y<-y[keep,]
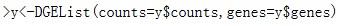
>y<-calcNormFactors(y) #默认为TMM标准化
3)检查样本的outlier and relationship
>y<-plotMDS(y) # 图1表明样本在两个维度都契合得很好
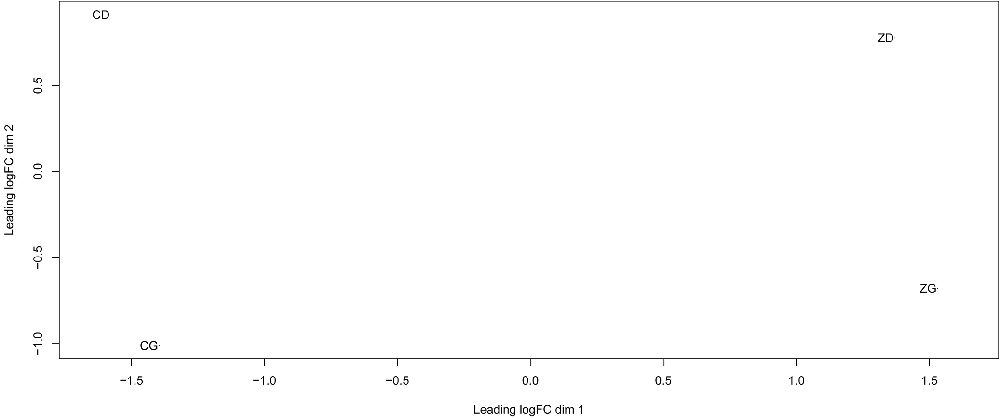 FIG.2
FIG.2
4)设计design matrix
>treat<-factor(c(‘d’,’g’,’d’,’g’))
>sample<-factor(c(‘b’,’b’,’m’,’m’))
>design<-model.matrix(~treat+sample)
>rownames(design)<-colnames(y)
5)推测dispersion
>y<-estimateGLMCommonDisp(y,design,verbose=TRUE)
>y<-estimateGLMTrendedDisp(y, design)
>y<-estimateGLMTagwiseDisp(y, design)
6)差异表达基因
>fit<-glmFit(y, design)
>lrt<-glmLRT(fit)
> topTags(lrt)#前10个差异表达基因
>summary(de<-decideTestsDGE(lrt))
>detags<-rownames(y)[as.logical(de)]
>plotSmear(lrt, de.tags=detags)
>abline(h=c(-1,1),col=’blue’) #蓝线为2倍差异表达基因,差异表达的数据在lrt中
 FIG.3
FIG.3
P.S. 由于时间仓促,如果文中有什么错误,请您跟我联系一起讨论,邮箱:
https://m.sciencenet.cn/blog-508298-776802.html
上一篇:分子系统发育树的节点支持率多少才可信?
下一篇:写文章的本质是什么?