博文
这个床上工具挺不错!
||
Bedtools是处理基因组信息分析的强大工具集合,本文列出自己学习其官方文档的几个点,对后面计算不同样品peak相似性的脚本做了下更新和调整,使用起来更为简单方便。
内容摘要
区域注释,如peak注释,peak分布分析,peak与调控元件交集等。
区域合并,如求算多样品peak合集,或合并重叠区域
区域互补,如得到非基因区
利用比对结果对测序广度和深度评估
多样品peak相似性计算,评估ChIP类区域结果的样品相似性。
bedtools主要功能
bedtools: flexible tools for genome arithmetic and DNA sequence analysis. usage: bedtools <subcommand> [options] The bedtools sub-commands include: [ Genome arithmetic ] intersect Find overlapping intervals in various ways. 求区域之间的交集,可以用来注释peak,计算reads比对到的基因组区域 不同样品的peak之间的peak重叠情况。 window Find overlapping intervals within a window around an interval. closest Find the closest, potentially non-overlapping interval. 寻找最近但可能不重叠的区域 coverage Compute the coverage over defined intervals. 计算区域覆盖度 map Apply a function to a column for each overlapping interval. genomecov Compute the coverage over an entire genome. merge Combine overlapping/nearby intervals into a single interval. 合并重叠或相接的区域 cluster Cluster (but don't merge) overlapping/nearby intervals. complement Extract intervals _not_ represented by an interval file. 获得互补区域 subtract Remove intervals based on overlaps b/w two files. 计算区域差集 slop Adjust the size of intervals. 调整区域大小,如获得转录起始位点上下游3 K的区域 flank Create new intervals from the flanks of existing intervals. sort Order the intervals in a file. 排序,部分命令需要排序过的bed文件 random Generate random intervals in a genome. 获得随机区域,作为背景集 shuffle Randomly redistrubute intervals in a genome. 根据给定的bed文件获得随机区域,作为背景集 sample Sample random records from file using reservoir sampling. spacing Report the gap lengths between intervals in a file. annotate Annotate coverage of features from multiple files. [ Multi-way file comparisons ] multiinter Identifies common intervals among multiple interval files. unionbedg Combines coverage intervals from multiple BEDGRAPH files. [ Paired-end manipulation ] pairtobed Find pairs that overlap intervals in various ways. pairtopair Find pairs that overlap other pairs in various ways. [ Format conversion ] bamtobed Convert BAM alignments to BED (& other) formats. bedtobam Convert intervals to BAM records. bamtofastq Convert BAM records to FASTQ records. bedpetobam Convert BEDPE intervals to BAM records. bed12tobed6 Breaks BED12 intervals into discrete BED6 intervals. [ Fasta manipulation ] getfasta Use intervals to extract sequences from a FASTA file. 提取给定位置的FASTA序列 maskfasta Use intervals to mask sequences from a FASTA file. nuc Profile the nucleotide content of intervals in a FASTA file. [ BAM focused tools ] multicov Counts coverage from multiple BAMs at specific intervals. tag Tag BAM alignments based on overlaps with interval files. [ Statistical relationships ] jaccard Calculate the Jaccard statistic b/w two sets of intervals. 计算数据集相似性 reldist Calculate the distribution of relative distances b/w two files. fisher Calculate Fisher statistic b/w two feature files. [ Miscellaneous tools ] overlap Computes the amount of overlap from two intervals. igv Create an IGV snapshot batch script. 用于生成一个脚本,批量捕获IGV截图 links Create a HTML page of links to UCSC locations. makewindows Make interval "windows" across a genome. 把给定区域划分成指定大小和间隔的小区间 (bin) groupby Group by common cols. & summarize oth. cols. (~ SQL "groupBy") 分组结算,不只可以用于bed文件。 expand Replicate lines based on lists of values in columns. split Split a file into multiple files with equal records or base pairs.
安装bedtools
ct@ehbio:~$ conda install bedtools
获得测试数据集(http://quinlanlab.org/tutorials/bedtools/bedtools.html)
ct@ehbio:~$ mkdir bedtools
ct@ehbio:~$ cd bedtools
ct@ehbio:~$ url=https://s3.amazonaws.com/bedtools-tutorials/web
ct@ehbio:~/bedtools$ curl -O ${url}/maurano.dnaseI.tgz
ct@ehbio:~/bedtools$ curl -O ${url}/cpg.bed
ct@ehbio:~/bedtools$ curl -O ${url}/exons.bed
ct@ehbio:~/bedtools$ curl -O ${url}/gwas.bed
ct@ehbio:~/bedtools$ curl -O ${url}/genome.txt
ct@ehbio:~/bedtools$ curl -O ${url}/hesc.chromHmm.bed交集 (intersect)

查看输入文件,bed格式,至少三列,分别是染色体,起始位置(0-based,
包括),终止位置
(1-based,不包括)。第四列一般为区域名字,第五列一般为空,第六列为链的信息。更详细解释见http://www.genome.ucsc.edu/FAQ/FAQformat.html#format1。
自己做研究CpG岛信息可以从UCSC的Table Browser获得,具体操作见http://blog.genesino.com/2013/05/ucsc-usages/。
ct@ehbio:~/bedtools$ head -n 3 cpg.bed exons.bed ==> cpg.bed <== chr1 28735 29810 CpG:_116 chr1 135124 135563 CpG:_30 chr1 327790 328229 CpG:_29 ==> exons.bed <== chr1 11873 12227 NR_046018_exon_0_0_chr1_11874_f 0 + chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 + chr1 13220 14409 NR_046018_exon_2_0_chr1_13221_f 0 +
获得重叠区域(既是外显子,又是CpG岛的区域)
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed | head -5 chr1 29320 29370 CpG:_116 chr1 135124 135563 CpG:_30 chr1 327790 328229 CpG:_29 chr1 327790 328229 CpG:_29 chr1 327790 328229 CpG:_29
输出重叠区域对应的原始区域(与外显子存在交集的CpG岛)
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -wa -wb > | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370
NR_024540_exon_10_0_chr1_29321_r 0 -chr1 135124 135563 CpG:_30 chr1 134772 139696
NR_039983_exon_0_0_chr1_134773_r 0 -chr1 327790 328229 CpG:_29 chr1 324438 328581
NR_028322_exon_2_0_chr1_324439_f 0 +chr1 327790 328229 CpG:_29 chr1 324438 328581
NR_028325_exon_2_0_chr1_324439_f 0 +chr1 327790 328229 CpG:_29 chr1 327035 328581
NR_028327_exon_3_0_chr1_327036_f 0 +
计算重叠碱基数
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -wo | head -10
chr1 28735 29810 CpG:_116 chr1 29320 29370
NR_024540_exon_10_0_chr1_29321_r 0 - 50chr1 135124 135563 CpG:_30 chr1 134772 139696
NR_039983_exon_0_0_chr1_134773_r 0 - 439chr1 327790 328229 CpG:_29 chr1 324438 328581
NR_028322_exon_2_0_chr1_324439_f 0 + 439chr1 327790 328229 CpG:_29 chr1 324438 328581
NR_028325_exon_2_0_chr1_324439_f 0 + 439chr1 327790 328229 CpG:_29 chr1 327035 328581
NR_028327_exon_3_0_chr1_327036_f 0 + 439chr1 713984 714547 CpG:_60 chr1 713663 714068
NR_033908_exon_6_0_chr1_713664_r 0 - 84chr1 762416 763445 CpG:_115 chr1 761585 762902
NR_024321_exon_0_0_chr1_761586_r 0 - 486chr1 762416 763445 CpG:_115 chr1 762970 763155
NR_015368_exon_0_0_chr1_762971_f 0 + 185chr1 762416 763445 CpG:_115 chr1 762970 763155
NR_047519_exon_0_0_chr1_762971_f 0 + 185chr1 762416 763445 CpG:_115 chr1 762970 763155
NR_047520_exon_0_0_chr1_762971_f 0 + 185
计算第一个(-a)bed区域有多少个重叠的第二个(-b)bed文件中有多少个区域
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -c | head chr1 28735 29810 CpG:_116 1 chr1 135124 135563 CpG:_30 1 chr1 327790 328229 CpG:_29 3 chr1 437151 438164 CpG:_84 0 chr1 533219 534114 CpG:_94 0 chr1 544738 546649 CpG:_171 0 chr1 713984 714547 CpG:_60 1 chr1 762416 763445 CpG:_115 10 chr1 788863 789211 CpG:_28 9
另外还有-v取出不重叠的区域,
-f限定重叠最小比例,-sorted可以对按sort -k1,1 -k2,2n排序好的文件加速操作。
同时对多个区域求交集 (可以用于peak的多维注释)
# -names标注注释来源 # -sorted: 如果使用了这个参数,提供的一定是排序好的bed文件 ct@ehbio:~/bedtools$ bedtools intersect -a exons.bed \ -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -names cpg gwas chromhmm \ | head -10000 | tail -10
chr1 27632676 27635124
NM_001276252_exon_15_0_chr1_27632677_chromhmm chr1 27633213
27635013 5_Strong_Enhancerchr1 27632676 27635124
NM_001276252_exon_15_0_chr1_27632677_chromhmm chr1 27635013
27635413 7_Weak_Enhancerchr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f
chromhmm chr1 27632613 27632813 6_Weak_Enhancerchr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f
chromhmm chr1 27632813 27633213 7_Weak_Enhancerchr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f
chromhmm chr1 27633213 27635013 5_Strong_Enhancerchr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f
chromhmm chr1 27635013 27635413 7_Weak_Enhancerchr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f cpg
chr1 27648453 27649006 CpG:_63chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f
chromhmm chr1 27648613 27649413 1_Active_Promoterchr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f cpg
chr1 27648453 27649006 CpG:_63chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f
chromhmm chr1 27648613 27649413 1_Active_Promoter
合并区域
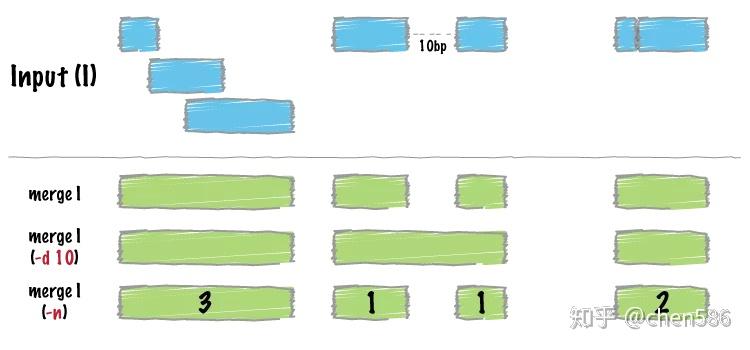
bedtools merge输入的是按sort -k1,1 -k2,2n排序好的bed文件。
只需要输入一个排序好的bed文件,默认合并重叠或邻接区域。
ct@ehbio:~/bedtools$ bedtools merge -i exons.bed | head -n 5 chr1 11873 12227 chr1 12612 12721 chr1 13220 14829 chr1 14969 15038 chr1 15795 15947
合并区域并输出此合并后区域是由几个区域合并来的
ct@ehbio:~/bedtools$ bedtools merge -i exons.bed -c 1 -o count | head -n 5 chr1 11873 12227 1 chr1 12612 12721 1 chr1 13220 14829 2 chr1 14969 15038 1 chr1 15795 15947 1
合并相距90 nt内的区域,并输出是由哪些区域合并来的
# -c: 指定对哪些列进行操作 # -o: 与-c对应,表示对指定列进行哪些操作 # 这里的用法是对第一列做计数操作,输出这个区域是由几个区域合并来的 # 对第4列做收集操作,记录合并的区域的名字,并逗号分隔显示出来 ct@ehbio:~/bedtools$ bedtools merge -i exons.bed -d 340 -c 1,4 -o count,collapse | head -4 chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f chr1 12612 12721 1 NR_046018_exon_1_0_chr1_12613_f chr1 13220 15038 3 NR_046018_exon_2_0_chr1_13221_f,NR_024540_exon_0_0_chr1_14362_r,NR_024540_exon_1_0_chr1_14970_r chr1 15795 15947 1 NR_024540_exon_2_0_chr1_15796_r
计算互补区域
给定一个全集,再给定一个子集,求另一个子集。比如给定每条染色体长度和外显子区域,求非外显子区域。给定基因区,求非基因区。给定重复序列,求非重复序列等。
重复序列区域的获取也可以用下面提供的链接:http://blog.genesino.com/2013/05/ucsc-usages/。
ct@ehbio:~/bedtools$ head genome.txt chr1 249250621 chr10 135534747 chr11 135006516 chr11_gl000202_random 40103 chr12 133851895 chr13 115169878 chr14 107349540 chr15 102531392 ct@ehbio:~/bedtools$ bedtools complement -i exons.bed -g genome.txt | head -n 5 chr1 0 11873 chr1 12227 12612 chr1 12721 13220 chr1 14829 14969 chr1 15038 15795
基因组覆盖广度和深度
计算基因组某个区域是否被覆盖,覆盖深度多少。有下图多种输出格式,也支持RNA-seq数据,计算junction-reads覆盖。
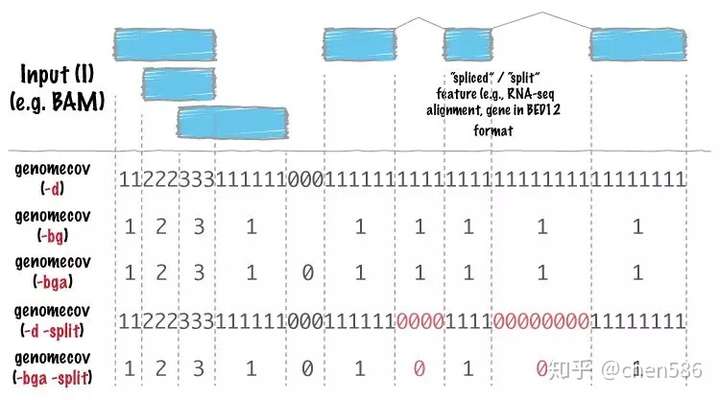
genome.txt里面的内容就是染色体及对应的长度。
# 对单行FASTA,可如此计算
# 如果是多行FASTA,则需要累加
ct@ehbio:~/bedtools$ awk 'BEGIN{OFS=FS="\t"}{\
if($0~/>/) {seq_name=$0;sub(">","",seq_name);} \
else {print seq_name,length;} }' ../bio/genome.fa | tee ../bio/genome.txt
chr1 60001
chr2 54001
chr3 54001
chr4 60001
ct@ehbio:~/bedtools$ bedtools genomecov -ibam ../bio/map.sortP.bam -bga \
-g ../bio/genome.txt | head
# 这个warning很有意思,因为BAM中已经有这个信息了,就不需要提供了
*****
*****WARNING: Genome (-g) files are ignored when BAM input is provided.
*****
# bedgraph文件,前3列与bed相同,最后一列表示前3列指定的区域的覆盖度。
chr1 0 11 0
chr1 11 17 1
chr1 17 20 2
chr1 20 31 3
chr1 31 36 4
chr1 36 43 6
chr1 43 44 7
chr1 44 46 8
chr1 46 48 9
chr1 48 54 10两个思考题:
怎么计算有多少基因组区域被测到了?
怎么计算平均测序深度是多少?
https://m.sciencenet.cn/blog-118204-1228868.html
上一篇:Cell|严景华/齐建勋首次发现新冠病毒进入细胞的详细分子细节
下一篇:来一场蛋白和小分子的风花雪月