博文
代码分析 | 单细胞转录组质控详解
||
前言
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
在前期处理单细胞转录组数据中,Quality Control(QC)是至关重要的一环(Hemberg-lab单细胞转录组数据分析(三)- 原始数据质控),直接关系到后期分析数据的可靠性。在单细胞转录组分析中,我们发现不同QC造成的结果也是略有差异,比如细胞周期、线粒体相关基因和核糖体相关基因等均会对下游数据分析造成影响,甚至我们使用多少的基因-cutoff值也会对我们的数据产生重要影响。比如我曾经看到的数据中出现的异种细胞的doublet使用双细胞筛选软件并没有进行有效的去除(单细胞预测Doublets软件包汇总-过渡态细胞是真的吗?),而使用cutoff值需要将基因数设置很高,以至于后期捕获的有效细胞数较少,不利于后期分析。
最近找到了芬兰CSC-IT科学中心主讲的生物信息课程(https://www.csc.fi/web/training/-/scrnaseq),
官网上还提供了练习素材以及详细代码,今天就来练习一下单细胞数据QC的过程:
第一部分是使用Seurat(https://satijalab.org/seurat/)来对QC进行可视化并进行细胞过滤;
第二部分将详细探讨scater软件包(https://bioconductor.org/packages/release/bioc/html/scater.html)。
加载环境
如果需要加载conda环境(软件安装不上,可能是网速慢!Conda/R/pip/brew等国内镜像大全拿走不谢~~),请在运行以下代码之前遵循以下说明:https://nbisweden.github.io/excelerate-scRNAseq/computing_environment_instructions.html。
数据集
在本教程中,我们将使用10x Genomics网站上的3个不同的PBMC数据集(https://support.10xgenomics.com/single-cell-gene-expression/datasets)。包括:
1k PBMCs using 10x v2 chemistry
1k PBMCs using 10x v3 chemistry
1k PBMCs using 10x v3 chemistry in combination with cell surface proteins, but disregarding the protein data and only looking at gene expression.
我们可以通过以下命令进行下载:
mkdir data #建立data文件夹 cd data curl -O http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v2/pbmc_1k_v2_filtered_feature_bc_matrix.h5 curl -O http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_filtered_feature_bc_matrix.h5 curl -O http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_protein_v3/pbmc_1k_protein_v3_filtered_feature_bc_matrix.h5
加载R包
suppressMessages(require(Seurat)) suppressMessages(require(scater)) suppressMessages(require(Matrix))
读入数据
v3.1k <- Read10X_h5("~/scrna-seq2019/day1/1-qc/pbmc_1k_v3_filtered_feature_bc_matrix.h5", use.names = T)
v2.1k <- Read10X_h5("~/scrna-seq2019/day1/1-qc/pbmc_1k_v2_filtered_feature_bc_matrix.h5", use.names = T)
p3.1k <- Read10X_h5("~/scrna-seq2019/day1/1-qc/pbmc_1k_protein_v3_filtered_feature_bc_matrix.h5", use.names = T)
# CITE-seq data中只提取基因表达矩阵
p3.1k <- p3.1k$`Gene Expression`Seurat
构建Seurat对象
首先,为每个数据集创建Seurat对象,然后合并为一个大的seurat对象。
sdata.v2.1k <- CreateSeuratObject(v2.1k, project = "v2.1k")
sdata.v3.1k <- CreateSeuratObject(v3.1k, project = "v3.1k")
sdata.p3.1k <- CreateSeuratObject(p3.1k, project = "p3.1k")
# 使用merge函数合并为一个seurat object. 为了防止数据集之间的barcodes重叠,给细胞添加ID。
alldata <- merge(sdata.v2.1k, c(sdata.v3.1k,sdata.p3.1k), add.cell.ids=c("v2.1k","v3.1k","p3.1k"))
# 在metadata中添加v2和v3标识。
chemistry <- rep("v3",ncol(alldata))
chemistry[Idents(alldata) == "v2.1k"] <- "v2"
alldata <- AddMetaData(alldata, chemistry, col.name = "Chemistry")
alldata
## An object of class Seurat
## 33538 features across 2931 samples within 1 assay
## Active assay: RNA (33538 features)
# 检查每个样本的细胞数量,将其存储在metadata的orig.ident中,并自动设置为active ident。
table(Idents(alldata))
##
## p3.1k v2.1k v3.1k
## 713 996 1222计算线粒体比例
Seurat可以自动计算一些QC统计信息,例如每个细胞的UMI数量和基因数分别存储在metadata的nCount_RNA和nFeature_RNA列(Hemberg-lab单细胞转录组数据分析(六)- 构建表达矩阵,UMI介绍)。
head(alldata@meta.data) ## orig.ident nCount_RNA nFeature_RNA Chemistry ## v2.1k_AAACCTGAGCGCTCCA-1 v2.1k 6631 2029 v2 ## v2.1k_AAACCTGGTGATAAAC-1 v2.1k 2196 881 v2 ## v2.1k_AAACGGGGTTTGTGTG-1 v2.1k 2700 791 v2 ## v2.1k_AAAGATGAGTACTTGC-1 v2.1k 3551 1183 v2 ## v2.1k_AAAGCAAGTCTCTTAT-1 v2.1k 3080 1333 v2 ## v2.1k_AAAGCAATCCACGAAT-1 v2.1k 5769 1556 v2
我们将手动计算线粒体reads的比例,并将其添加到metadata中。
mt.genes <- rownames(alldata)[grep("^MT-",rownames(alldata))]
C<-GetAssayData(object = alldata, slot = "counts")
percent.mito <- colSums(C[mt.genes,])/Matrix::colSums(C)*100
alldata <- AddMetaData(alldata, percent.mito, col.name = "percent.mito")计算核糖体比例
我们用上面一样的方法来计算源自核糖体蛋白的基因表达比例。
rb.genes <- rownames(alldata)[grep("^RP[SL]",rownames(alldata))]
percent.ribo <- colSums(C[rb.genes,])/Matrix::colSums(C)*100
alldata <- AddMetaData(alldata, percent.ribo, col.name = "percent.ribo")Plot QC
绘制小提琴图展示一些QC指标(feature)(可视化之为什么要使用箱线图?):
VlnPlot(alldata, features = "nFeature_RNA", pt.size = 0.1) + NoLegend()

VlnPlot(alldata, features = "nCount_RNA", pt.size = 0.1) + NoLegend()

VlnPlot(alldata, features = "percent.mito", pt.size = 0.1) + NoLegend()

VlnPlot(alldata, features = "percent.ribo", pt.size = 0.1) + NoLegend()

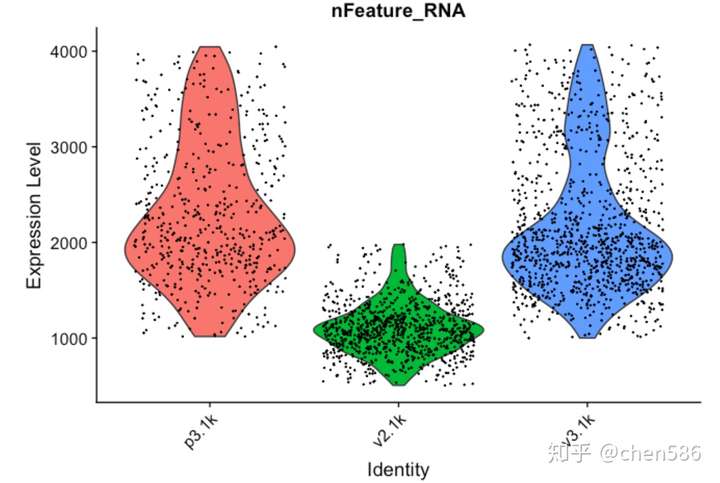
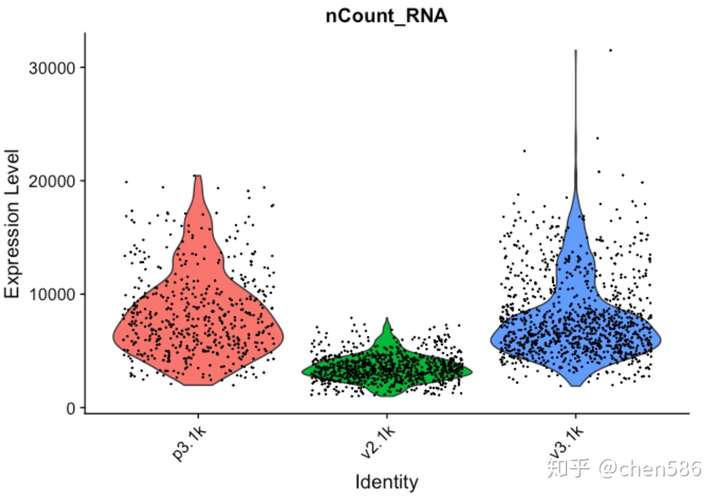
如图所示,v2检测到的基因数较低,但检测出的核糖体蛋白比例却偏高。检测到较少的低表达基因是因为核糖体蛋白发生高表达,使得核糖体基因在转录景观中占较大比例。
QC方式不同时的散点图:
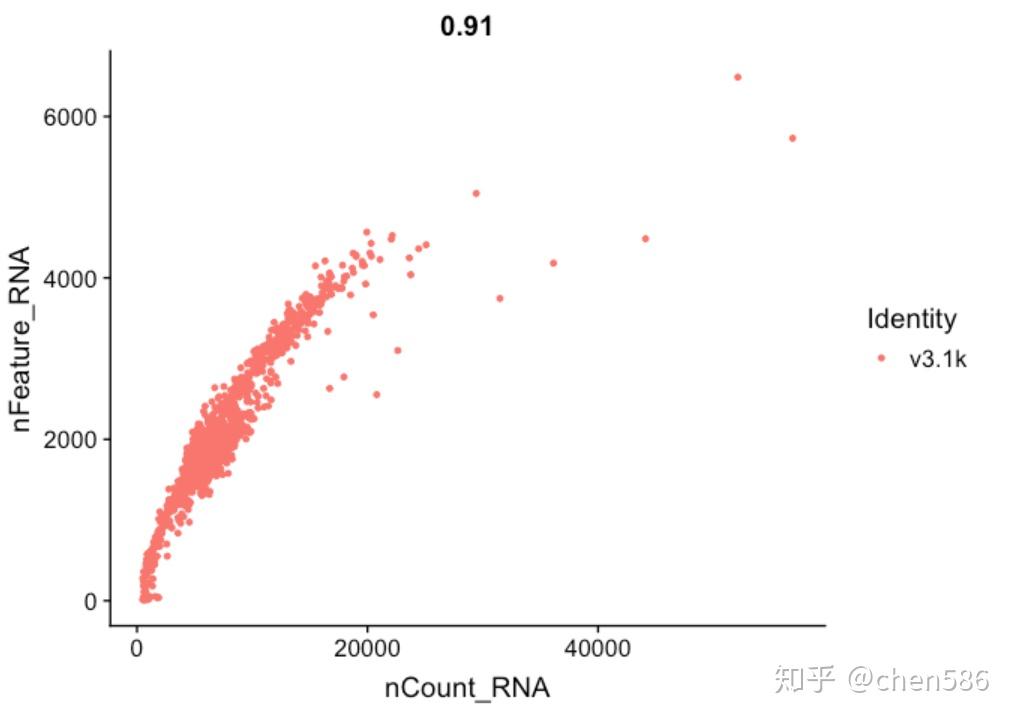
FeatureScatter(alldata, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")

FeatureScatter(alldata, feature1 = "nFeature_RNA", feature2 = "percent.mito")

FeatureScatter(alldata, feature1="percent.ribo", feature2="nFeature_RNA")

我们还可以展示一个数据集:
FeatureScatter(alldata, feature1 = "nCount_RNA", feature2 = "nFeature_RNA", cells = WhichCells(alldata, expression = orig.ident == "v3.1k") )
过滤
线粒体过滤
线粒体reads比例高的细胞有很多,如果过滤掉它们后还剩下足够的细胞,那删除这些细胞可能是较为明智的。另一个选择是从数据集中删除所有线粒体reads,并希望剩余的基因仍然具有足够的生物学信号。第三种选择是在标准化过程中去除percent.mito变量。
在此次示例中,某些细胞中的线粒体reads比例高达99.7%,细胞信号很好的可能性不高,因此去除这些细胞。
可以通过展示的图决定合理的cutoff值。在这次例子中,大部分细胞的线粒体reads低于25%,可以将其用作临界值。
# 选择线粒体基因百分比<25%的细胞 selected <- WhichCells(alldata, expression = percent.mito < 25) length(selected) ## [1] 2703 # 对细胞取子集 data.filt <- subset(alldata, cells = selected) # 画小提琴图 VlnPlot(data.filt, features = "percent.mito")

如图所示,mito百分比仍有很大差异,因此必须在数据分析步骤中进行处理。
基因过滤
检测到的基因数量高的离谱的话可能表明doublets,也有可能是因为样品中某一种细胞具有较多基因表达。
在这次的数据集中,v2和v3之间在基因检测方面也存在明显差异,因此对所有数据应用相同的临界值可能并不合适。
同时,在蛋白质测定数据p3.1k中,存在大量细胞几乎没有检测到具有双峰分布的基因。v3.1k和v2.12个数据集中也看不到这种分布类型。考虑到它们都是pbmc数据集,因此可以将这种分布视为低质量的库。
通过设置cutoff值分别为4100(对于v3 chemistry)和2000(对于v2)过滤测到高基因的(可能的doublets)细胞。
#从检测到许多基因的细胞开始。 high.det.v3 <- WhichCells(data.filt, expression = nFeature_RNA > 4100) high.det.v2 <- WhichCells(data.filt, expression = nFeature_RNA > 2000 & orig.ident == "v2.1k") # 细胞取子集 data.filt <- subset(data.filt, cells=setdiff(WhichCells(data.filt),c(high.det.v2,high.det.v3))) # 查看细胞数 ncol(data.filt) ## [1] 2631
用低基因检测(低质量文库)过滤细胞,其中v3的基因数cutoff为1000,v2的基因数cutoff为500。
#筛选基因 low.det.v3 <- WhichCells(data.filt, expression = nFeature_RNA < 1000 & orig.ident != "v2.1k") low.det.v2 <- WhichCells(data.filt, expression = nFeature_RNA < 500 & orig.ident == "v2.1k") # 去除细胞 data.filt <- subset(data.filt, cells=setdiff(WhichCells(data.filt),c(low.det.v2,low.det.v3))) # 查看细胞数 ncol(data.filt) ## [1] 2531
再次绘制QC图
VlnPlot(data.filt, features = "nFeature_RNA", pt.size = 0.1) + NoLegend()
VlnPlot(data.filt, features = "nCount_RNA", pt.size = 0.1) + NoLegend()
VlnPlot(data.filt, features = "percent.mito", pt.size = 0.1) + NoLegend()

# 在过滤前后检查每个样本的细胞数 table(Idents(alldata)) ## ## p3.1k v2.1k v3.1k ## 713 996 1222 table(Idents(data.filt)) ## ## p3.1k v2.1k v3.1k ## 526 933 1072
计算细胞周期分数
Seurat具有根据已知的S期和G2/M期基因列表计算细胞周期分数的功能(Seurat亮点之细胞周期评分和回归)。
data.filt <- CellCycleScoring(
object = data.filt,
g2m.features = cc.genes$g2m.genes,
s.features = cc.genes$s.genes
)
VlnPlot(data.filt, features = c("S.Score","G2M.Score"))
从上图可以看出不同数据集中处于S期和G2M期的细胞Score基本均为0,表明细胞周期对数据质量影响并不大。
Scater
可以使用scater包完成相似的QC绘图和细胞过滤,但是由于我们使用Seurat筛选了细胞,因此现在仅使用scater探索数据中的技术偏差。
可以直接从count矩阵创建SCE对象,网址为:https://rdrr.io/bioc/scater/f/vignettes/overview.Rmd
在该例中,我们可以直接从Seurat对象转换为SCE对象。
sce <- as.SingleCellExperiment(data.filt)
我们用访问函数来评估SingleCellExperiment对象的不同元素。
counts(object):用于返回read计数矩阵。如上所示,如果未为该对象定义任何计数,则计数矩阵slot为
NULL。exprs(object):用于返回(对数计数)表达式值的矩阵,实际上为访问对象的对数计数slot(
logcounts的同义词)。
SCE对象还有的slot:
细胞元数据(
Cell metadata,),可以以DataFrame提供,其中行是细胞,列是细胞属性(例如细胞类型、培养条件、捕获的天数等)。特征元数据(
Feature metadata),可以以DataFrame提供,其中行是特征(例如基因),列是特征属性,例如Ensembl ID,生物型,gc含量等。
计算QC指标
默认情况下,QC指标是根据count data计算得出的。但是可以通过exprs_values参数进行更改。我们还可以在函数调用中包加入有关哪些基因是线粒体的信息。
# 计算QC指标 sce <- calculateQCMetrics(sce, feature_controls = list(mito = mt.genes)) # check what all entries are - colnames(colData(sce)) ## [1] "orig.ident" ## [2] "nCount_RNA" ## [3] "nFeature_RNA" ## [4] "Chemistry" ## [5] "percent.mito" ## [6] "percent.ribo" ## [7] "S.Score" ## [8] "G2M.Score" ## [9] "Phase" ## [10] "ident" ## [11] "is_cell_control" ## [12] "total_features_by_counts" ## [13] "log10_total_features_by_counts" ## [14] "total_counts" ## [15] "log10_total_counts" ## [16] "pct_counts_in_top_50_features" ## [17] "pct_counts_in_top_100_features" ## [18] "pct_counts_in_top_200_features" ## [19] "pct_counts_in_top_500_features" ## [20] "total_features_by_counts_endogenous" ## [21] "log10_total_features_by_counts_endogenous" ## [22] "total_counts_endogenous" ## [23] "log10_total_counts_endogenous" ## [24] "pct_counts_endogenous" ## [25] "pct_counts_in_top_50_features_endogenous" ## [26] "pct_counts_in_top_100_features_endogenous" ## [27] "pct_counts_in_top_200_features_endogenous" ## [28] "pct_counts_in_top_500_features_endogenous" ## [29] "total_features_by_counts_feature_control" ## [30] "log10_total_features_by_counts_feature_control" ## [31] "total_counts_feature_control" ## [32] "log10_total_counts_feature_control" ## [33] "pct_counts_feature_control" ## [34] "pct_counts_in_top_50_features_feature_control" ## [35] "pct_counts_in_top_100_features_feature_control" ## [36] "pct_counts_in_top_200_features_feature_control" ## [37] "pct_counts_in_top_500_features_feature_control" ## [38] "total_features_by_counts_mito" ## [39] "log10_total_features_by_counts_mito" ## [40] "total_counts_mito" ## [41] "log10_total_counts_mito" ## [42] "pct_counts_mito" ## [43] "pct_counts_in_top_50_features_mito" ## [44] "pct_counts_in_top_100_features_mito" ## [45] "pct_counts_in_top_200_features_mito" ## [46] "pct_counts_in_top_500_features_mito"
如您所见,scater为细胞计算了许多不同的QC指标。
Scater还可以根据基因计算一些统计信息:
colnames(rowData(sce)) ## [1] "is_feature_control" "is_feature_control_mito" ## [3] "mean_counts" "log10_mean_counts" ## [5] "n_cells_by_counts" "pct_dropout_by_counts" ## [7] "total_counts" "log10_total_counts"
绘制QC统计数据
高变基因:
让我们看看表达的前50位基因是什么。
plotHighestExprs(sce, exprs_values = "counts")

如图所示,MALAT1对应平均约4%的计数。在某些细胞中,一些count高达〜30%,可以考虑在进一步分析和聚类之前删除该基因。而且,线粒体基因在总计数中所占比例很高的话也应该被删除。
累积表达:
绘制文库大小的相对比例,该比例由每个细胞的最高表达基因(默认为500个基因)表示。这可以帮助我们寻找样本之间表达分布的差异。
# 绘制每个样本 plotScater(sce, block1 = "ident", nfeatures = 1000)

绘制基因统计:
函数plotRowData可以绘制rowData中的任何统计信息,例如均值表达与检测到的细胞数。
plotRowData(sce, x = "n_cells_by_counts", y = "mean_counts")

绘制细胞统计:
用函数plotColData可以以相同的方式绘制细胞的任何qc统计分析。
p1 <- plotColData(sce, x = "total_counts", y = "total_features_by_counts", colour_by = "ident") p2 <- plotColData(sce, x = "pct_counts_feature_control", y = "total_features_by_counts", colour_by = "ident") p3 <- plotColData(sce, x = "pct_counts_feature_control", y = "pct_counts_in_top_50_features", colour_by = "ident") multiplot(p1, p2, p3, cols = 2)

识别QC统计数据中的异常值
识别低质量细胞的方法是在所有qc-stats上运行PCA,然后在PCA空间中识别异常值(Hemberg-lab单细胞转录组数据分析(十一)- Scater单细胞表达谱PCA可视化)。
sce <- runPCA(sce, use_coldata = TRUE, detect_outliers = TRUE) ## sROC 0.1-2 loaded plotReducedDim(sce, use_dimred="PCA_coldata", colour_by = "ident")
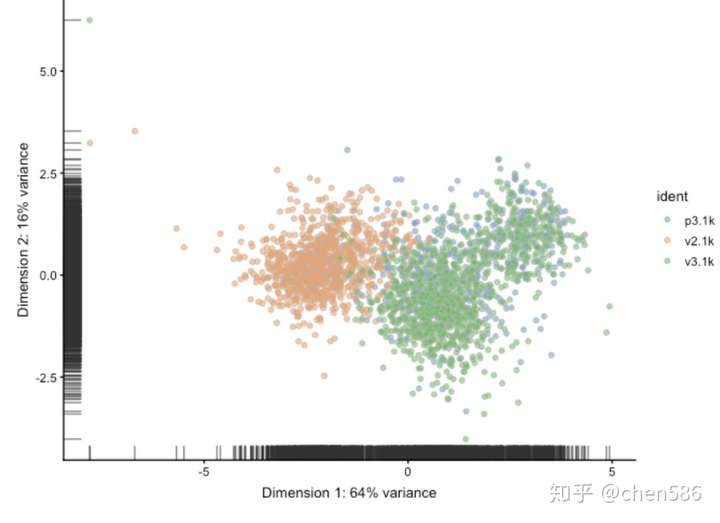
#检查是否有异常值 table(colData(sce)$outlier) ## ## FALSE ## 2531
在这种情况下,我们已经滤除了低质量的细胞,并且在QC PCA中未检测到任何异常值。
降维
建议先用runPCA、runTSNE等函数做降维( 用了这么多年的PCA可视化竟然是错的!!!),以便将它们存储在SCE对象中,这样就不必在每次绘制时都重新运行这些函数。除了这两个函数可以做降维,还有其他函数,比如plotPCA和plotTSNE等等,或者使用函数plotReducedDim并指定use_dimred = "pca"等类似的。
在使用任意组分信息进行降维之前,请记住要设置随机种子,以便可以完全复现。
# 使用前1000高变基因运行PCA sce <- runPCA(sce, ntop = 1000, exprs_values = "logcounts", ncomponents = 20) # PCA - with different coloring, first 4 components # first by sample plotPCA(sce,ncomponents=4,colour_by="ident")

# then by Celltype plotPCA(sce,ncomponents=4,colour_by="percent.mito")
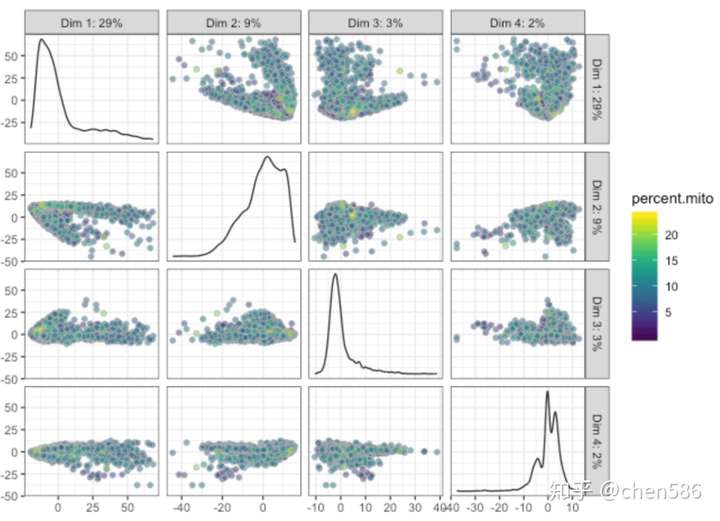
# Diffusion map, 运行需要安装destiny包! set.seed(1) sce <- runDiffusionMap(sce, ntop = 1000, ncomponents = 4) plotDiffusionMap(sce, colour_by="ident",ncomponents=4)

# tSNE -使用tSNE对降维数据进行可视化,top10PCs set.seed(1) sce <- runTSNE(sce, ntop = 1000, ncomponents = 2, perplexity = 30, n_dimred = 10) plotTSNE(sce, colour_by="ident")

# UMAP,需要安装umap包! set.seed(1) sce <- runUMAP(sce) plotUMAP(object = sce, colour_by="ident")

影响因子的可视化
我们可以使用plotExplanatoryVariables函数调查不同因素的相对重要性。当拟合线性模型将每个基因的表达值与该因子进行回归时,我们计算colData(sce)中每个因子的可决系数R2来评估拟合优度,最好是在对数表达式值上执行此操作,可以减少平均值对方差的影响——因此,我们首先要进行归一化(取log值)。
下面的可视化计算耗时较长。
plotExplanatoryVariables(sce, variables = c("ident","Chemistry","pct_counts_mito", "total_features_by_counts", "pct_counts_in_top_50_features", "pct_counts_in_top_500_features", "total_counts", "S.Score","G2M.Score"))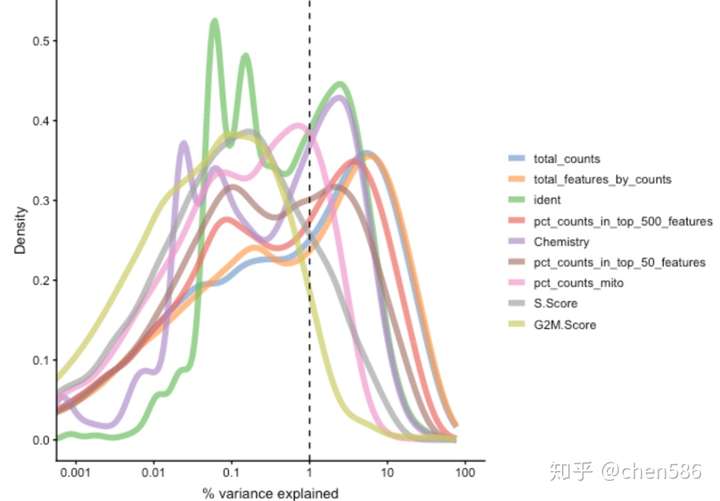
每条线对应一个因子在所有基因中R平方值的分布。
我们要留意确定与QC或Metadata密切相关的PC。默认情况下是绘制前10个因子,但是在这里我们仅可视化一些特定因子。
# for total_features
plotExplanatoryPCs(sce, variables = c("ident", "Chemistry","pct_counts_mito", "total_features_by_counts", "pct_counts_in_top_50_features", "total_counts","S.Score","G2M.Score"), npcs_to_plot = 20)
问题:您认为可以在数据中看到一些明显的批次效应吗?您是否认为存在技术偏差?
PC1显然与数据的分布相关,例如前50-500个高度表达的基因在计数的比例,以及占total_features/total_counts的比例。这是scRNAseq数据中的常见问题,可能是技术偏差,或者是大小和转录方式非常不同的细胞类型的生物学特征造成的。
同样很明显的是,许多topPC(尤其是PC 2、3、6、7、8)在很大程度上是由不同的样本(ident)或仅由v2 vs v3(Chemistry)来解释的。
Session info
sessionInfo() ## R version 3.5.1 (2018-07-02) ## Platform: x86_64-apple-darwin13.4.0 (64-bit) ## Running under: macOS 10.14.4 ## ## Matrix products: default ## BLAS/LAPACK: /Users/asbj/Programs/miniconda3_4.2.12/envs/elixir-course/lib/R/lib/libRblas.dylib ## ## locale: ## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8 ## ## attached base packages: ## [1] parallel stats4 stats graphics grDevices utils datasets ## [8] methods base ## ## other attached packages: ## [1] Matrix_1.2-17 scater_1.10.1 ## [3] ggplot2_3.1.1 SingleCellExperiment_1.4.0 ## [5] SummarizedExperiment_1.12.0 DelayedArray_0.8.0 ## [7] BiocParallel_1.16.6 matrixStats_0.54.0 ## [9] Biobase_2.42.0 GenomicRanges_1.34.0 ## [11] GenomeInfoDb_1.18.1 IRanges_2.16.0 ## [13] S4Vectors_0.20.1 BiocGenerics_0.28.0 ## [15] Seurat_3.0.0 ## ## loaded via a namespace (and not attached): ## [1] reticulate_1.12 R.utils_2.8.0 ## [3] tidyselect_0.2.5 htmlwidgets_1.3 ## [5] grid_3.5.1 trimcluster_0.1-2.1 ## [7] ranger_0.11.1 Rtsne_0.15 ## [9] munsell_0.5.0 destiny_2.12.0 ## [11] codetools_0.2-16 ica_1.0-2 ## [13] umap_0.2.0.0 future_1.12.0 ## [15] sROC_0.1-2 withr_2.1.2 ## [17] colorspace_1.4-1 knitr_1.20 ## [19] ROCR_1.0-7 robustbase_0.93-2 ## [21] vcd_1.4-4 VIM_4.8.0 ## [23] TTR_0.23-4 gbRd_0.4-11 ## [25] listenv_0.7.0 Rdpack_0.10-1 ## [27] labeling_0.3 GenomeInfoDbData_1.2.1 ## [29] cvTools_0.3.2 bit64_0.9-7 ## [31] rhdf5_2.26.2 ggthemes_4.1.1 ## [33] diptest_0.75-7 R6_2.4.0 ## [35] ggbeeswarm_0.6.0 robCompositions_2.0.10 ## [37] rsvd_1.0.0 RcppEigen_0.3.3.5.0 ## [39] mvoutlier_2.0.9 hdf5r_1.2.0 ## [41] flexmix_2.3-15 bitops_1.0-6 ## [43] reshape_0.8.8 assertthat_0.2.1 ## [45] SDMTools_1.1-221.1 scales_1.0.0 ## [47] nnet_7.3-12 beeswarm_0.2.3 ## [49] gtable_0.3.0 npsurv_0.4-0 ## [51] globals_0.12.4 rlang_0.3.4 ## [53] scatterplot3d_0.3-41 splines_3.5.1 ## [55] lazyeval_0.2.2 yaml_2.2.0 ## [57] reshape2_1.4.3 abind_1.4-5 ## [59] tools_3.5.1 zCompositions_1.2.0 ## [61] gplots_3.0.1.1 RColorBrewer_1.1-2 ## [63] proxy_0.4-22 ggridges_0.5.1 ## [65] Rcpp_1.0.1 plyr_1.8.4 ## [67] zlibbioc_1.28.0 purrr_0.3.2 ## [69] RCurl_1.95-4.12 pbapply_1.4-0 ## [71] viridis_0.5.1 cowplot_0.9.4 ## [73] zoo_1.8-5 haven_2.1.0 ## [75] ggrepel_0.8.1 cluster_2.0.9 ## [77] magrittr_1.5 RSpectra_0.14-0 ## [79] data.table_1.12.2 openxlsx_4.1.0 ## [81] lmtest_0.9-36 RANN_2.6 ## [83] truncnorm_1.0-8 mvtnorm_1.0-10 ## [85] fitdistrplus_1.0-14 hms_0.4.2 ## [87] lsei_1.2-0 evaluate_0.13 ## [89] smoother_1.1 rio_0.5.16 ## [91] mclust_5.4.3 readxl_1.3.1 ## [93] gridExtra_2.3 compiler_3.5.1 ## [95] tibble_2.1.1 KernSmooth_2.23-15 ## [97] crayon_1.3.4 R.oo_1.22.0 ## [99] htmltools_0.3.6 pcaPP_1.9-73 ## [101] tidyr_0.8.3 rrcov_1.4-4 ## [103] MASS_7.3-51.4 fpc_2.1-11.1 ## [105] boot_1.3-22 car_3.0-2 ## [107] sgeostat_1.0-27 R.methodsS3_1.7.1 ## [109] gdata_2.18.0 metap_1.1 ## [111] igraph_1.2.4.1 forcats_0.4.0 ## [113] pkgconfig_2.0.2 foreign_0.8-71 ## [115] laeken_0.5.0 sp_1.3-1 ## [117] plotly_4.8.0 vipor_0.4.5 ## [119] XVector_0.22.0 bibtex_0.4.2 ## [121] NADA_1.6-1 stringr_1.4.0 ## [123] digest_0.6.18 sctransform_0.2.0 ## [125] pls_2.7-0 tsne_0.1-3 ## [127] rmarkdown_1.11 cellranger_1.1.0 ## [129] DelayedMatrixStats_1.4.0 curl_3.3 ## [131] kernlab_0.9-27 gtools_3.8.1 ## [133] modeltools_0.2-22 nlme_3.1-139 ## [135] jsonlite_1.6 Rhdf5lib_1.4.3 ## [137] carData_3.0-2 viridisLite_0.3.0 ## [139] pillar_1.3.1 lattice_0.20-38 ## [141] GGally_1.4.0 httr_1.4.0 ## [143] DEoptimR_1.0-8 survival_2.44-1.1 ## [145] xts_0.11-1 glue_1.3.1 ## [147] zip_2.0.1 png_0.1-7 ## [149] prabclus_2.2-7 bit_1.1-14 ## [151] class_7.3-15 stringi_1.4.3 ## [153] HDF5Array_1.10.1 caTools_1.17.1.2 ## [155] dplyr_0.8.0.1 irlba_2.3.3 ## [157] e1071_1.7-0 future.apply_1.1.0 ## [159] ape_5.3
撰文:Tiger
编辑:生信宝典
https://m.sciencenet.cn/blog-118204-1228874.html
上一篇:来一场蛋白和小分子的风花雪月
下一篇:代码分析 | 单细胞转录组Normalization详解