博文
代码分析 | 单细胞转录组clustering详解
||
聚类可视化
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
我们在单细胞转录组分析中最为常用的聚类可视化即为tSNE和UMAP(Hemberg-lab单细胞转录组数据分析(十二)- Scater单细胞表达谱tSNE可视化),不过非线性可视化方法(例如t-SNE)通常会扰乱数据中的全局结构。diffusion maps能够很好的表达局部和全局结构,但无法针对二维(2D)可视化进行优化,因为它们不是专门为可视化设计的。(其实即便如此,我现在也习惯性应用diffusion maps,可以说明更多的信息,虽然有时候确实是很难看。。。)
芬兰CSC-IT科学中心主讲的生物信息课程(https://www.csc.fi/web/training/-/scrnaseq)视频,官网上还提供了练习素材以及详细代码,今天就来练习一下单细胞数据clustering的过程。
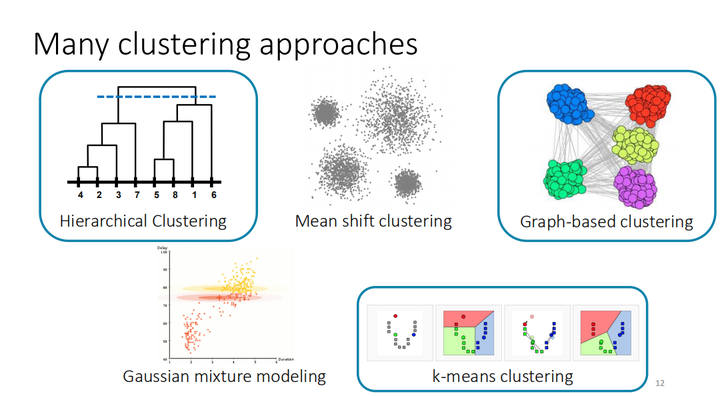
在本教程中,我们将研究不同的聚类scRNA-seq数据集方法以表征不同的细胞亚群。
所需加载包
suppressMessages(require(tidyverse)) suppressMessages(require(Seurat)) suppressMessages(require(cowplot)) suppressMessages(require(scater)) suppressMessages(require(scran)) suppressMessages(require(igraph))
数据集
本教程使用的是来自发育中的小鼠胚胎的小细胞数据集[1]。该数据集已进行了预处理、创建了SingleCellExperiment对象并对细胞进行了注释(cellassign:用于肿瘤微环境分析的单细胞注释工具(9月Nature))。
数据下载链接:https://github.com/NBISweden/excelerate-scRNAseq/tree/master/session-clustering/session-clustering_files。
#加载表达矩阵
deng <- readRDS("session-clustering_files/deng-reads.rds")
deng
#查看细胞类型注释 table(colData(deng)$cell_type2)

特征选择
第一步是决定在细胞聚类中使用哪些基因(Hemberg-lab单细胞转录组数据分析(十)- Scater基因评估和过滤)。单细胞RNA-seq可以分析细胞中的大量基因,大多数基因的表达不足以提供有意义的信号并且通常受到技术噪音的干扰,其中可能会添加一些不必要的信号,从而使生物变异模糊,而基因过滤还可以加快下游分析的计算时间。
首先看一下所有基因的表达平均值和方差。哪些基因似乎不太重要,哪些可能是技术噪音?
#计算整个细胞的基因平均值 gene_mean <- rowMeans(counts(deng)) #计算整个细胞的基因方差 gene_var <- rowVars(counts(deng)) #ggplot plot library(tidyverse) gene_stat_df <- tibble(gene_mean,gene_var) ggplot(data=gene_stat_df ,aes(x=log(gene_mean), y=log(gene_var))) + geom_point(size=0.5) + theme_classic()
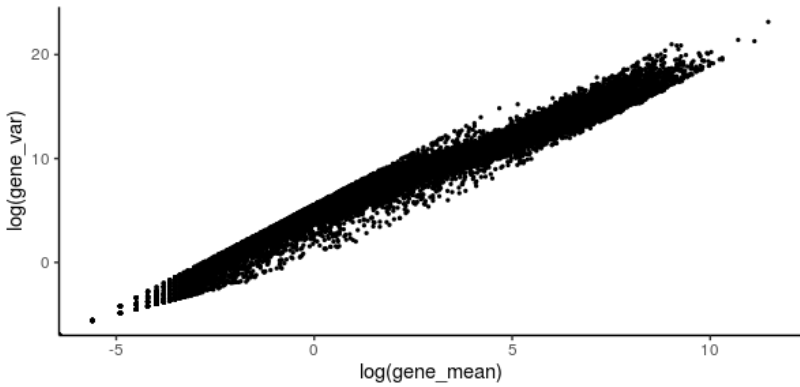
滤除低丰度基因
低丰度基因大多无信息,不能代表数据的生物学差异。它们通常是由技术噪音(例如dropout)导致,在下游分析中的存在通常会导致准确性降低,因为它们会干扰将要使用的某些统计模型,并且会毫无意义地增加计算时间,这在处理非常大的数据时可能至关重要。
abundant_genes <- gene_mean >= 0.5 #去除低丰度基因 # 绘制基因表达分布 hist(log10(gene_mean), breaks=100, main="", col="grey80", xlab=expression(Log[10]~"average count")) abline(v=log10(0.5), col="red", lwd=2, lty=2)
#在SingleCellExperiment Object中去除低丰度基因 deng <- deng[abundant_genes,] dim(deng)

过滤在很少细胞中表达的基因
我们还可以过滤少量细胞中的某些基因。此过程将删除一些在一两个细胞中高度表达的异常基因。这些基因不需要进一步分析,因为它们主要来自人为的不规则扩增。值得注意的是,当分析的目的是检测数据中非常稀有的亚群时,我们可能不希望进行此过程。
#计算每个非零表达基因的数量 numcells <- nexprs(deng, byrow=TRUE) #过滤不到5个细胞中检测到的基因 numcells2 <- numcells >= 5 deng <- deng[numcells2,] dim(deng)
检测高变的基因
highly variable gene(HVG)假设基因在细胞之间的表达差异很大,则其中的一些差异是由于细胞之间的生物学差异而不是技术噪音引起的。但是,基因的平均表达与不同细胞reads计数的差异之间存在正相关关系。如果仅保留高变异基因,将保留许多在每个细胞中表达但不能代表生物学变异的高表达管家基因。因此为了正确识别HVG,必须纠正此关系。
我们可以使用下列任一方法来确定高变基因。
选项1 :将变异系数建模为平均值的函数。
# out <- technicalCV2(deng, spike.type=NA, assay.type= "counts") # out$genes <- rownames(deng) # out$HVG <- (out$FDR<0.05) # as_tibble(out) # # # 绘制高变基因 # ggplot(data = out) + geom_point(aes(x=log2(mean), y=log2(cv2), color=HVG), size=0.5) + geom_point(aes(x=log2(mean), y=log2(trend)), color="red", size=0.1) # # ## 将HVG存入metadata # metadata(deng)$hvg_genes <- rownames(deng)[out$HVG]
选项2 :根据平均数对生物成分的方差建模。
首先,我们估算每个基因表达的差异,然后将差异分解为生物学和技术成分。然后将HVGs鉴定为具有最大生物学成分的基因。这避免了优先排序对由于技术因素(例如在RNA捕获和文库制备过程中的采样噪声)而高度可变的基因。详细内容查看scran vignette:https://bioconductor.org/packages/devel/bioc/vignettes/scran/inst/doc/scran.html#5_variance_modelling
fit <- trendVar(deng, parametric=TRUE, use.spikes = FALSE) dec <- decomposeVar(deng, fit) dec$HVG <- (dec$FDR<0.00001) hvg_genes <- rownames(dec[dec$FDR < 0.00001, ]) # plot highly variable genes plot(dec$mean, dec$total, pch=16, cex=0.6, xlab="Mean log-expression", ylab="Variance of log-expression") o <- order(dec$mean) lines(dec$mean[o], dec$tech[o], col="dodgerblue", lwd=2) points(dec$mean[dec$HVG], dec$total[dec$HVG], col="red", pch=16)
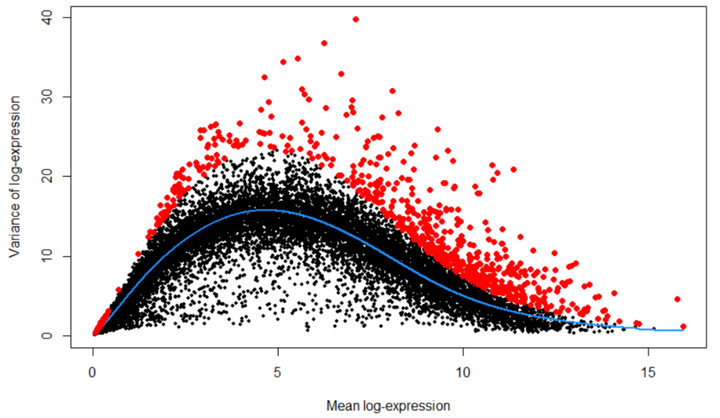
## save the decomposed variance table and hvg_genes into metadata for safekeeping metadata(deng)$hvg_genes <- hvg_genes metadata(deng)$dec_var <- dec
降维
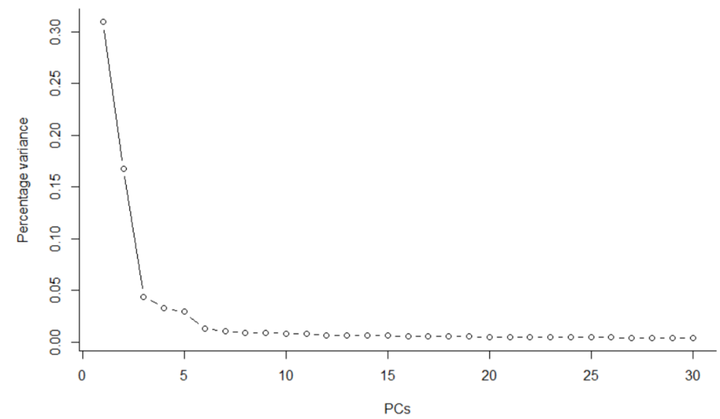
由于高频率的噪声(技术和生物学的)和大量的维度(即基因)导致聚类在计算上具有一定的困难,因此需要通过应用降维方法(例如PCA、tSNE和UMAP)解决这些问题。
#PCA (选择组分数量) deng <- runPCA(deng, method = "irlba", ncomponents = 30, feature_set = metadata(deng)$hvg_genes) #绘制不同PC对变异解释的差异 X <- attributes(deng@reducedDims$PCA) plot(X$percentVar~c(1:30), type="b", lwd=1, ylab="Percentage variance" , xlab="PCs" , bty="l" , pch=1)
画PCA plot(PC1 vs. PC2) :
plotReducedDim(deng, "PCA", colour_by = "cell_type2")
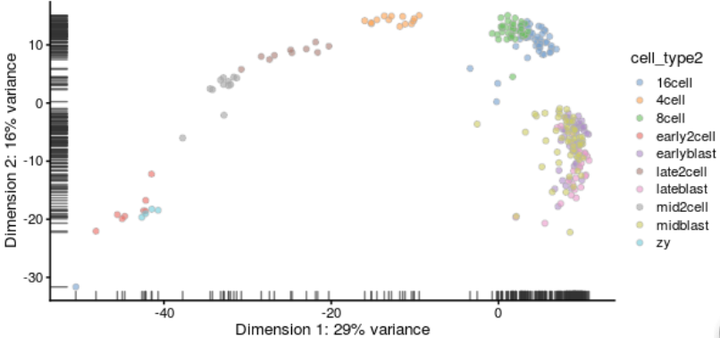
绘制tSNE图时需要注意tSNE是随机方法。每次运行它时都会得到略有不同的结果。为了方便起见可以设置随机种子,这样就可以获得相同的结果。
#tSNE deng <- runTSNE(deng, perplexity = 30, feature_set = metadata(deng)$hvg_genes, set.seed = 1) plotReducedDim(deng, "TSNE", colour_by = "cell_type2")
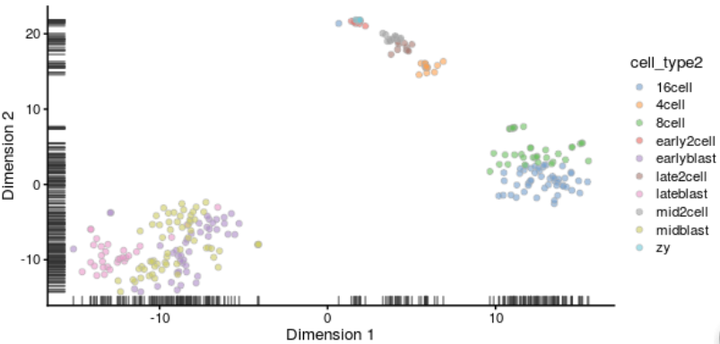
聚类
层次聚类
#计算距离(默认值:欧几里得距离) distance_eucledian <- dist(t(logcounts(deng))) #使用ward linkage执行分层聚类 ward_hclust_eucledian <- hclust(distance_eucledian,method = "ward.D2") plot(ward_hclust_eucledian, main = "dist = eucledian, Ward linkage")
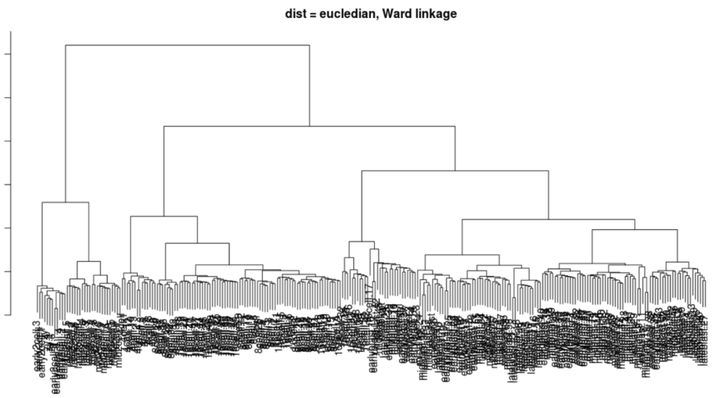
我反正是啥也看不见。。。但是看起来层次分类蛮明显的。。。
现在,设置树状图参数生成10个亚群,并在PCA图上绘制聚类标签。
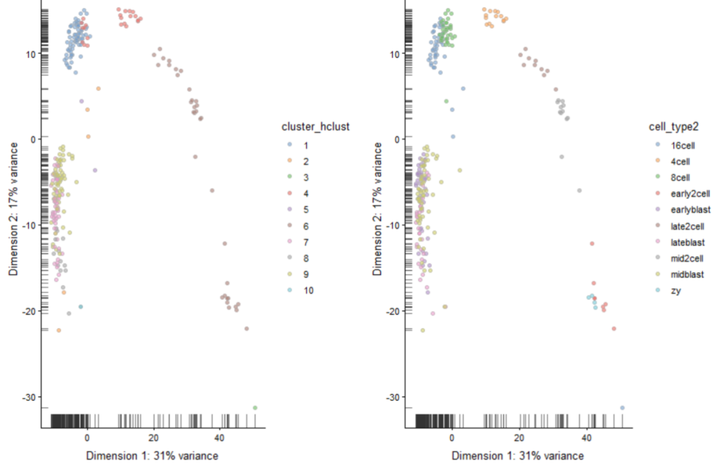
#设置树状图参数生成10个亚群 cluster_hclust <- cutree(ward_hclust_eucledian,k = 10) colData(deng)$cluster_hclust <- factor(cluster_hclust) plot_grid(plotReducedDim(deng, "PCA", colour_by = "cluster_hclust"), plotReducedDim(deng, "PCA", colour_by = "cell_type2"))
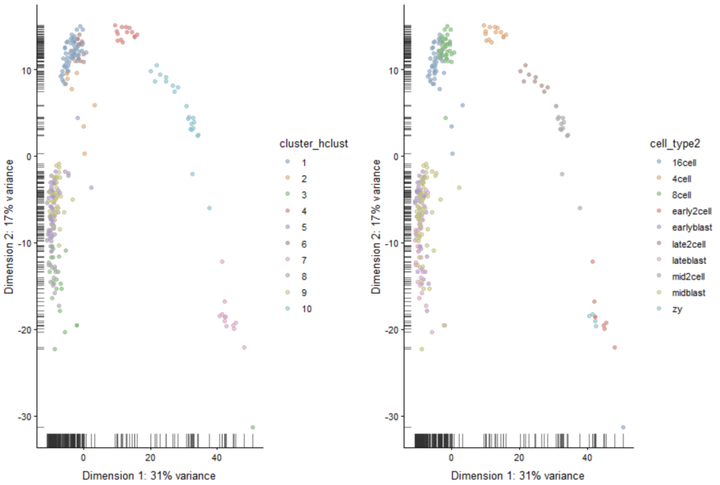
在tSNE图上绘制聚类标签。
plot_grid(plotReducedDim(deng, "TSNE", colour_by = "cluster_hclust"), plotReducedDim(deng, "TSNE", colour_by = "cell_type2"))
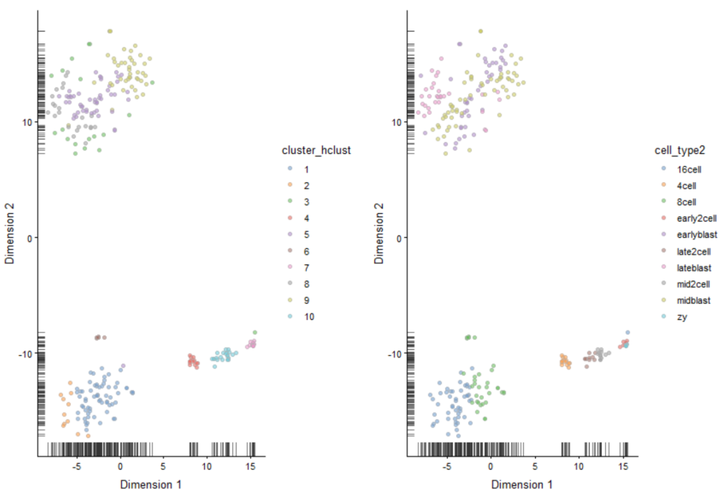
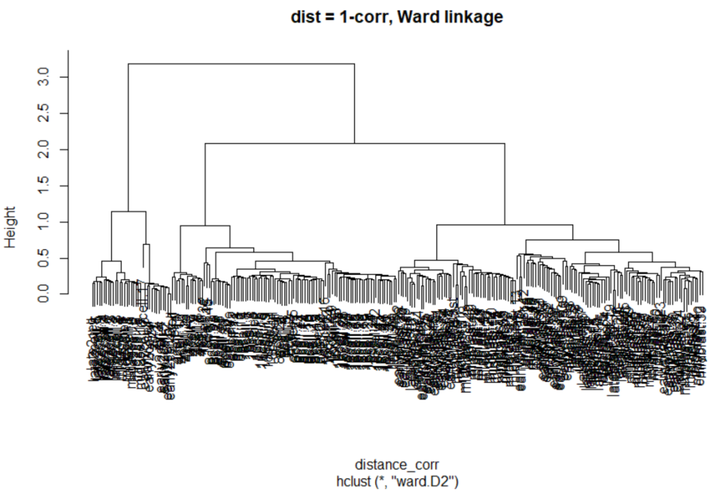
我们尝试另一种距离参数。常用的距离参数是1-correlation。
# 计算距离(1 - correlation) C <- cor(logcounts(deng)) # Run clustering based on the correlations, where the distance will # be 1-correlation, e.g. higher distance with lower correlation. distance_corr <- as.dist(1-C) #使用ward linkage进行分层聚类 ward_hclust_corr <- hclust(distance_corr,method="ward.D2") plot(ward_hclust_corr, main = "dist = 1-corr, Ward linkage")
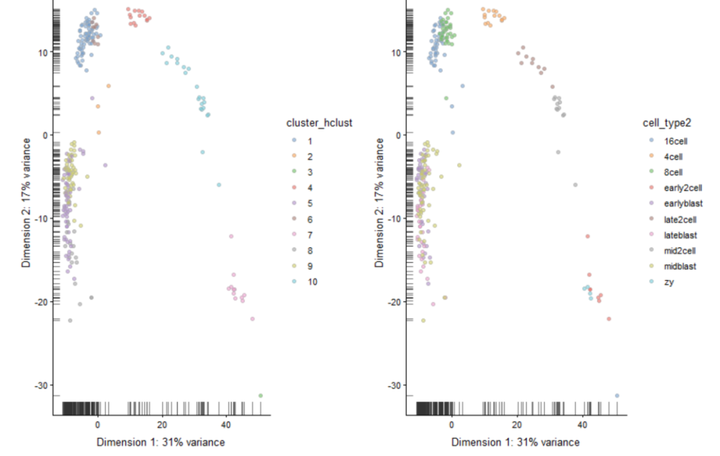
再次设置树状图参数生成10个亚群,并在PCA图上绘制聚类标签。
#设置树状图参数生成10个亚群 cluster_hclust <- cutree(ward_hclust_corr,k = 10) colData(deng)$cluster_hclust <- factor(cluster_hclust) plot_grid(plotReducedDim(deng, "PCA", colour_by = "cluster_hclust"), plotReducedDim(deng, "PCA", colour_by = "cell_type2"))
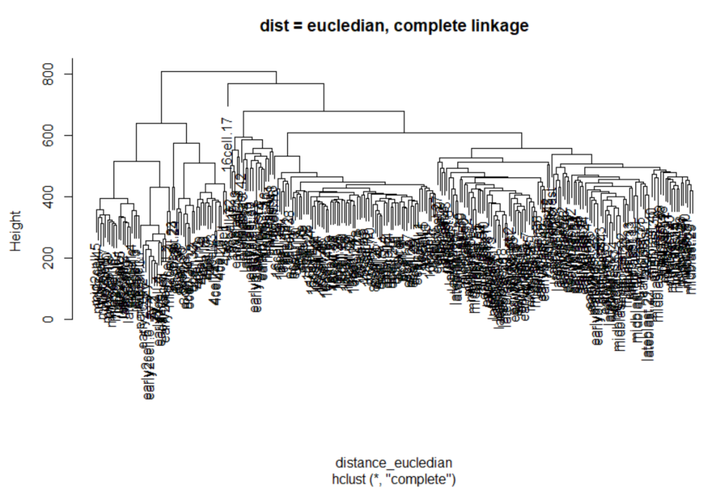
除了更改距离度量,我们还可以更改链接方法 —— 使用完全链接,而不是使用Ward的方法。
# 计算距离 (default: Eucledian distance) distance_eucledian <- dist(t(logcounts(deng))) #使用ward linkage进行分层聚类 comp_hclust_eucledian <- hclust(distance_eucledian,method = "complete") plot(comp_hclust_eucledian, main = "dist = eucledian, complete linkage")
再一次,切割树状图以生成10个细胞亚群,并在PCA图上绘制亚群标签。
#设置树状图参数以生成10个细胞亚群 cluster_hclust <- cutree(comp_hclust_eucledian,k = 10) colData(deng)$cluster_hclust <- factor(cluster_hclust) plot_grid(plotReducedDim(deng, "PCA", colour_by = "cluster_hclust"), plotReducedDim(deng, "PCA", colour_by = "cell_type2"))
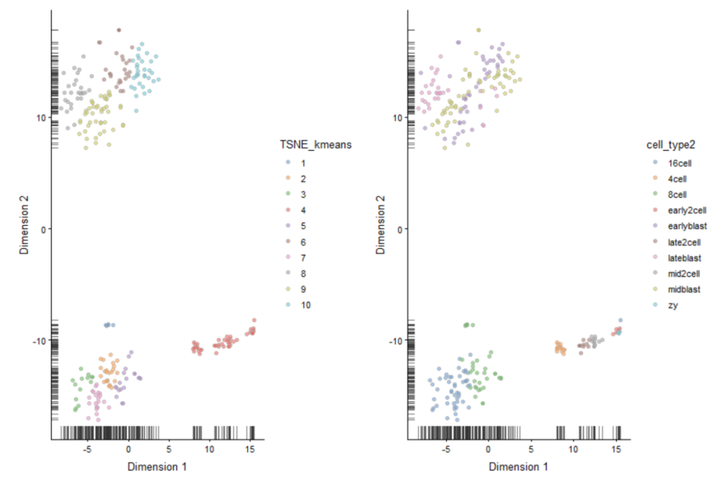
tSNE + Kmeans
# 在tSNE坐标上执行kmeans算法 deng_kmeans <- kmeans(x = deng@reducedDims$TSNE,centers = 10) TSNE_kmeans <- factor(deng_kmeans$cluster) colData(deng)$TSNE_kmeans <- TSNE_kmeans #Compare with ground truth plot_grid(plotTSNE(deng, colour_by = "TSNE_kmeans"), plotTSNE(deng, colour_by = "cell_type2"))
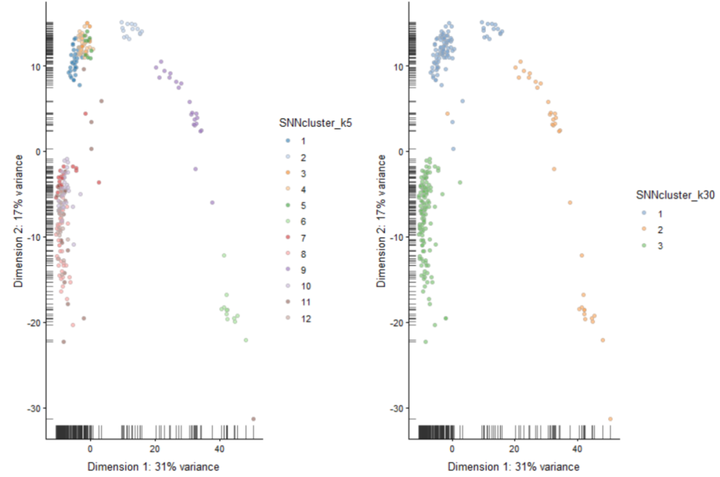
Graph Based Clustering
#k=5 #The k parameter defines the number of closest cells to look for each cells SNNgraph_k5 <- buildSNNGraph(x = deng, k=5) SNNcluster_k5 <- cluster_louvain(SNNgraph_k5) colData(deng)$SNNcluster_k5 <- factor(SNNcluster_k5$membership) p5<- plotPCA(deng, colour_by="SNNcluster_k5")+ guides(fill=guide_legend(ncol=2)) # k30 SNNgraph_k30 <- buildSNNGraph(x = deng, k=30) SNNcluster_k30 <- cluster_louvain(SNNgraph_k30) colData(deng)$SNNcluster_k30 <- factor(SNNcluster_k30$membership) p30 <- plotPCA(deng, colour_by="SNNcluster_k30") #plot the different clustering. plot_grid(p5+ guides(fill=guide_legend(ncol=1)),p30)
Session info
sessionInfo()## R version 3.5.3 (2019-03-11) ## Platform: x86_64-w64-mingw32/x64 (64-bit) ## Running under: Windows 10 x64 (build 17763) ## ## Matrix products: default ## ## locale: ## [1] LC_COLLATE=English_United States.1252 ## [2] LC_CTYPE=English_United States.1252 ## [3] LC_MONETARY=English_United States.1252 ## [4] LC_NUMERIC=C ## [5] LC_TIME=English_United States.1252 ## ## attached base packages: ## [1] parallel stats4 stats graphics grDevices utils datasets ## [8] methods base ##
[1]:https://science.sciencemag.org/content/343/6167/193
撰文:Tiger 校对:生信宝典
https://m.sciencenet.cn/blog-118204-1228881.html
上一篇:代码分析 | 单细胞转录组数据整合详解
下一篇:RNA-seq最强综述名词解释&思维导图|关于RNA-seq,你想知道的都在这(续)