ВЉЮФ
АыЕМЬхвьжЪНсФмДјЖдЦыНщЩм/Introduction about semiconductor band alignment
|||
зїепЃКZhang Zhaofu
вдЯТЪЧЮвдФЖСЮФЯзЁЂНсКЯздМКЕФОбщзіЕФвЛаЉЙигкАыЕМЬхвьжЪНсФмДјЖдЦыЕФзмНсНщЩмЃЌШчЙћгаШЮКЮВЛЖдЃЌЛЖгМАЪБСЊЯЕЮвзіаоИФЁЃЯЃЭћзЊдизЂУїдДГіДІЃКhttp://blog.sciencenet.cn/blog-2686986-1172373.html
ЕБАыЕМЬх/АыЕМЬхЃЌЛђепОјдЕЬх/АыЕМЬхНгДЅаЮГЩНчУцНсЙЙЪБ,вђЮЊНћДјПэЖШВЛЭЌЃЌдкСНВрВФСЯЕФЕМДјЕзКЭМлДјЖЅДІЛсаЮГЩВЛСЌајЕФЬЈНзЃЌМДФмДјЬЈНз(band offset)ЃЌЦфжаЕМДјЕзДІЕФФмДјЬЈНзБЛГЦЮЊЕМДјЬЈНз(conduction band offset, CBO)ЃЌМлДјЖЅДІЕФФмДјЬЈНзБЛГЦЮЊМлДјЬЈНз(valence band offset, VBO)ЁЃЪгФмДјЖдЦфЧщПіВЛЭЌЃЌАыЕМЬхвьжЪНсБЛЗжЮЊШ§РрЃКtype-I, type-II,КЭtype-IIIЃЌЦфФмДјЭМШчЭМ1[1]ЃК
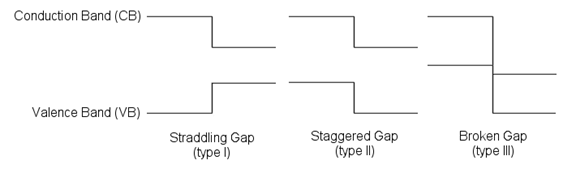
ЭМ1 The three types of semiconductor heterojunctions organized by band alignment.
АыЕМЬхФмДјЖдЦыгаживЊвтвхЁЃБШШчЃЌЖдгкЕчзгЦїМўРДЫЕЃЌГЃгУЕФЮЊtype-I band alignment, вЊЧѓCBO/VBOДѓгк1eVВХФмИќгааЇЕФЪјИПЕчзг/ПебЈЃЌМѕаЁТЉЕч[2]ЁЃtype-II вьжЪНсдквьжЪНсЙтДпЛЏЗжНтЫЎСьгђЃЌПЩвдгааЇМѕаЁЕчзгПебЈЖдЗћКЯЃЌЬсИпЙтДпЛЏаЇТЪ[3]ЁЃ
НчУцСНВрЕФФмДјЖдЦыЃЈband alignment)вРРЕгкСНВрВФСЯЕФЕчКЩзЊвЦЁЃЕБвьжЪНсСНВрЮоЕчКЩзЊвЦЪБКђЃЌCBOЪЧгЩвьжЪНсСНВрВФСЯЕФЕчзгЧзКЭЪЦ(electron affinity, EA)ОіЖЈЃЌБЛГЦЮЊelectron affinity modelЃЌгжГЦЮЊAnderson's rule[4]ЁЃдкетИіФЃаЭжаЃЌШЯЮЊСНВрецПеФмМЖГжЦНЃЌИљОнСНВрВФСЯЕФEAВЛЭЌЃЌЕУЕНСНВрВФСЯЕФCBЮЛжУЃЌМДCBOЁЃИљОнВФСЯЕФНћДјПэЖШEgЪ§жЕЃЌМДПЩЕУЕНМлДјЕФЯрЖдЙиЯЕЃЌМДVBOЁЃСэЭтЕФАьЗЈЪЧВЩгУвЛИіЭтРДЕФВЮПМФмМЖзіВЮПМБъзМЃЌБШШчВЩШЁЙ§ЖЩН№ЪєдгжЪЕФИпЖШОжгђЛЏЕФdЙьЕРФмМЖРДзіВЮПМБъзМ[5] ЃЌЛђепВЩгУВФСЯжаHЕФ(+/-)ФмМЖРДзіВЮПМБъзМ[6]ЁЃ
ЪЕМЪЕФНчУцжаЃЌЭЈГЃДцдкЕчКЩзЊвЦЃЌаЮГЩНчУцdipoleЁЃНчУцdipoleЛсЕїећband alignment, ЪЙЕУband alignment ВЛдйзёбelectron affinity modelЁЃУшЪігаЕчКЩзЊвЦЕФвьжЪНсЕФЮяРэФЃаЭгаЖрИіЃЌЦфжаГЃгУЕФCNLФЃаЭБэДяЪНЮЊ[2]ЃК
ЦфжаІжЮЊelectron affinityЃЌɸCNL charge neutrality level (CNL) ЪЧАыЕМЬхФмДјНсЙЙжаЕФвЛИіВЮПМФмМЖЃЌгЩВФСЯаджЪЃЈФмДјНсЙЙЃЉРДОіЖЈЁЃSЮЊFermi-level pinning factorЃЌБэеїАыЕМЬхВФСЯЕФFermi-level pinning ЬиЕуЕФЃКS=1БэеїЮоpinningЕФSchottky limitЃЛS=0БэеїЧПpinningЕФBardeen limitЁЃЖдгкАыЕМЬхвьжЪНсРДЫЕЃЌSЭЈГЃгЩEgИќДѓЕФвЛВрРДОіЖЈЁЃЖдКмЖрАыЕМЬхВФСЯБШШчSiРДЫЕЃЌSжЕБШНЯаЁЃЌЩЯЪіЙЋЪНЕкШ§ЯюПЩвдКіТдЃЌМДдкЧПpinningЧщПіЯТЃЌФмДјЖдЦыгЩСНВрВФСЯЕФCNLЕФЖдЦыРДШЗЖЈЁЃЖјCNLгыецПеФмМЖФмСПВюПЩвдБэеїАыЕМЬхВФСЯЕФЕчИКадЃЌМДЩЯЪіЙЋЪНБэЪОЃЌАыЕМЬхвьжЪНсЕФФмДјЖдЦыЪЧПМТЧSвђзгЦСБЮзїгУЯТвьжЪНсСНВрВФСЯЕФЕчИКаджЎВюЁЃЖдCNLФЃаЭЕФИќЯъЯИЬжТлПЩвдВЮПМ[2]ЁЃетРяднВЛЖдЦфЫћЬжТлАыЕМЬхвьжЪНсФмДјЖдЦыЕФЮяРэФЃаЭеЙПЊЯъЯИУшЪіЃЌгааЫШЄЕФПЩвдВЮПМ [7-8]вдМАИќЖрЕФЯрЙиЮФеТЁЃ
дкВФСЯМЦЫужаЃЌПЩвдЭЈЙ§НЈСЂАыЕМЬхвьжЪНсНчУцФЃаЭЃЌНјааЕквЛаддРэМЦЫуЕУЕНЁЃЙигкАыЕМЬхвьжЪНсНчУцНЈФЃЗНЗЈЕШгаЪБМфдйЕЅЖРПЊЬљНщЩмЁЃзЂвтЃЌВЛНЈСЂНчУцФЃаЭвВПЩвдМЦЫуВФСЯЕФband alignmentЃЌЕЋЪЧБиаывЊЧѓвЛИіВЮПМФмМЖРДНјааline-up.МДБуШчДЫЃЌетбљЧѓНтГіРДЕФband alignmentВЂУЛгаПМТЧЕНЕчКЩзЊвЦЕФаЇЙћЁЃ
НчУцФЃаЭжаШчКЮЕУЕНband alignment, гаЖрИіАьЗЈЁЃЦфжаНЯЮЊГЃМћЕФЮЊЃКlocal DOS (LDOS, or partial DOS, PDOS) methodЁЂcore-level alignment method, КЭ averaged potential alignment methodЁЃЭЈЙ§ЗжЮіМЦЫуЪ§ОнБуПЩЕУЕНVBO, ЖјЛёШЁCBOАьЗЈЃЌПЩвдИљОнЙЋЪНCBO=Eg1-Eg2-VBOЕУЕНЁЃНсКЯТлЮФЃЈНщЩмЧАСНИіmethodЕФЭМРДздЮвЕФТлЮФЃЌЛЖгв§гУЃЉЃЌж№вЛНщЩмШчЯТЃК
ЃЈ1ЃЉLDOS (or PDOS) method

ЭМ2 LDOS method to get the band alignment at HfO2/GaN interface.[9]
вдЩЯЭМЮвЗЂБэЕФТлЮФHfO2/GaNНчУцФЃаЭЮЊР§[9]НщЩмЁЃЪзЯШНЈСЂвьжЪНсНчУцФЃаЭЃЈЙигкАыЕМЬхвьжЪНсНчУцНЈФЃЗНЗЈЕШгаЪБМфдйЕЅЖРПЊЬљНщЩмЃЉЃЌШЛКѓЖдГкдЅЕФНчУцФЃаЭНјааDOSМЦЫуЁЃШЁНчУцСНВрдЖРыНчУцЕФGaNдзгКЭHfO2дзгЃЈвВПЩвдЗжБ№ШЁGaдзгNдзгHfдзгOдзгЃЉЛГіЦфPDOSЃЌетЪБЕУЕНЕФМлДјЖЅФмСПжЎВюМДЮЊVBOЁЃИУЗНЗЈдРэЮЊЃКдЖРыНчУцЕФдзгПЩвдНЯЩйЪмЕНinterface interactionЃЌМДЛљБОЩЯБЃГжЦфbulk behaviorЃЛШЯЮЊдкНчУцГЌАћФЃаЭжаСНВрВФСЯЕФецПеФмМЖЪЧГжЦНЕФЃЌЫљвдЕУЕНЕФМлДјЖЅФмСПжЎВюМДЮЊНчУцЕФVBOЁЃЖјЛёШЁCBOАьЗЈЃЌПЩвдИљОнЙЋЪНCBO=Eg1-Eg2-VBOЕУЕНЁЃзЂвт1ЃЌШчЙћЪЧгУМЦЫуЕФEgжЕдђБиаыПМТЧDFTМЦЫув§ШыЕФЕЭЙРНћДјЕФЮЪЬтЃЌМДВЩгУдгЛЏЗККЏМЦЫуЃЛвВПЩвдПМТЧжБНгШЁЪЕбщEgжЕЁЃзЂвт2ЃЌШчЙћЪЧгУИУЗНЗЈРДБШНЯВЛЭЌФЃаЭжаBOБфЛЏЧїЪЦЪЧзуЙЛЕФЃЌШчЙћЪЧЮЊСЫжБНгЕУЕНОпЬхжЕЃЌЛсДцдквЛЖЈЮѓВюЁЃ
ЃЈ2ЃЉCore-level alignment method

ЭМ3 Core level alignment method to get the band alignment at MoS2/GaN interface.[10]
вдЩЯЭМЮвЗЂБэЕФТлЮФMoS2/GaNНчУцФЃаЭЮЊР§[10]НщЩмЁЃетИіЗНЗЈЕФдРэЪЧЃКШЯЮЊЮоТлдкНчУцВФСЯЛЙЪЧЬхВФСЯжаЃЌВФСЯЕФМлДјЖЅгыаОФмМЖФмСПжЎВюЪЧЖЈжЕЁЃКЫЭтЕчзгЗжЮЊМлЕчзгКЭаОЕчзг(valence electrons and core electrons)ЃЌЦфжаcore electronгыcore stateБЛШЯЮЊЪЧЮШЖЈЕФЬЌЃЌВЛБЛГЩМќЕШРДгАЯьЁЃЪЕбщНЧЖШЃЌаОФмМЖПЩвдЭЈЙ§XPSБэеїЗНЗЈЕУЕНЃЌЫљвдИљОнетИіАьЗЈЃЌМШПЩвдгУгкЪЕбщбаОПвьжЪНсФмДјЖдЦыЃЌвВПЩвдЭЈЙ§МЦЫуРДЬжТлЁЃЩЯЭМЪЧЮвгУЪЕбщXPSЗНЗЈЕУЕНЕФMoS2/GaNвьжЪНсФмДјЖдЦыНсЙћЁЃдкЮФЯз[10]Ињ[11]жаЮввВДгМЦЫуНЧЖШЬсШЁСЫMoS2/GaNвдМАSiNx/GaNЕФФмДјЖдЦыЃЌИњЪЕбщжЕНЯЯрНќЁЃ
ЛљгкИУЗНЗЈзіМЦЫуЃЈЛђепЪЕбщЃЉЬжТлЕФОпЬхзіЗЈЃК1. МЦЫуЬхВФСЯAЃЈгыЬхВФСЯBЃЉЕФаОФмМЖЃЌЖСШЁГіаОФмМЖИњМлДјЖЅФмСПВюІЄEA (ІЄEB)ЃЛ2.ЬсШЁНчУцФЃаЭСНВрдЖРыНчУцЧјгђЕФдзгЕФаОФмМЖEcore-A (Ecore-B)ЃЛ3. Ecore-A+ ІЄEAгыEcore-B+ ІЄEB дђЮЊвьжЪНсСНВрABВФСЯИїздЕФМлДјЖЅЃЌЦфФмСПВюдђЮЊVBOЁЃИУЗНЗЈзМШЗадИќИпЃЌВЂЧвПЩвдгыЪЕбщНсКЯЃЌБЛКмЖрШЫВЩгУЁЃЭЦМіМИЦЊгУcore levelЕФКмОЕфЕФЮФЯз[12]ЁЃжСгкШчКЮДгМЦЫуНЧЖШЛёШЁаОФмМЖаХЯЂЃЌПЩвдПМТЧСНИіЗНЗЈЃКa. VASPМЦЫуINCARжаМгШыICORELEVEL=1МДПЩдкOUTCARжаЪфГіУПвЛИідзгЕФаОФмМЖаОФмМЖаХЯЂЁЃb.жБНгПМТЧдкdosЛђепbandЭМжаЖСГізюЩюЕФвЛИіband ЃЈor PDOSЃЉЕФФмСПЮЛжУзїЮЊcore level (or semi-core level)ЁЃ
ЃЈ3ЃЉAveraged potential alignment method
ИУЗНЗЈЮвднЪБЛЙЮДгУЙ§ЁЃЦфдРэИњcore-level methodНгНќЃЌШЯЮЊЃКШЯЮЊЮоТлдкНчУцВФСЯЛЙЪЧЬхВФСЯжаЃЌВФСЯЕФМлДјЖЅгыЦНОљОВЕчЪЦФмСПжЎВюЪЧЖЈжЕЁЃИУЗНЗЈЕФзіЗЈЮЊЃК1.ЕУЕНЬхВФСЯAЃЈЬхВФСЯBЃЉЕФplanar-averaged electrostatic potentialЃЛ2.ЖдЦфдйзіЦНОљЕУЕНmacroscopic-averaged electrostatic potentialЃЌЖСГіИУpotentialИњМлДјЖЅФмСПВюІЄEA (ІЄEB)ЃЛ3.ЬсШЁНчУцФЃаЭСНВрдЖРыНчУцЧјгђЕФМИИідзгВуЃЌЖСШЁГіЦфmacroscopic-averaged electrostatic potentialЗжБ№ЮЊ EA (EB)ЃЛ4.EA+ ІЄEAгыEB+ ІЄEB дђЮЊвьжЪНсСНВрABВФСЯИїздЕФМлДјЖЅЃЌЦфФмСПВюдђЮЊVBOЁЃКмШнвзПДГіЃЌИУЗНЗЈИњcore-levelЗНЗЈКмРрЫЦЁЃЦфгХЕуЪЧПЩвдНшжњaverage potentialЬжТлcharge transferЃЌЕЋЪЧШБЕуЪЧaveraged potentialетИіИХФюЪЧДПЮяРэЕФИХФюЃЌЪЕбщЕФВЮПМадНЯЕЭЁЃЭЦМіМИЦЊгУaveraged potential alignmentЕФКмОЕфЕФЮФЯзБШШч[13]ЁЃ
вдЩЯБуЪЧЮвдФЖСЮФЯзЁЂНсКЯздМКЕФОбщзіЕФвЛаЉЙигкАыЕМЬхвьжЪНсФмДјЖдЦыЕФзмНсНщЩмЃЌШчЙћгаШЮКЮВЛЖдЃЌЛЖгМАЪБСЊЯЕЮвзіаоИФЁЃСэЭтЛЖгв§гУЮвЕФЮФеТЃЌМДЬћзгжаЬсМАЕФ[9-11].ЯЃЭћзЊдизЂУїдДГіДІЃКhttp://blog.sciencenet.cn/blog-2686986-1172373.html
ВЙГфвЛИіТЉЕєЕФПЩвдНјааДжТдНјааФмДјЖдЦыЕФЗНЗЈЃЌcommon-anion rule: compound semiconductors which have the same anion will form an interface with near zero valence-band discontinuity [14]
[1] https://en.wikipedia.org/wiki/Heterojunction
[2] J. Robertson, J. Vac. Sci. Technol. B 18, 1785 (2000); J. Robertson, J. Vac. Sci. Technol. A, 31, 050821 (2013); J. Robertson, Journal of Applied Physics 100, 014111 (2006);
[3] R. Marschall, Adv. Funct. Mater. 2014, 24, 2421ЈC2440
[4] R. L. Andersen, Solid-State Electron. 5, 341 (1962).
[5] M. J. Caldas, A. Fazzio, and Alex Zunger, Appl. Phys. Lett. 45, 671 (1984).
[6] C. G. Van de Walle, and J. Neugebauer, Nature 423, 626ЈC628 (2003)
[7] J. O. McCaldin,T. C. McGill, and C. A. Mead, Phys. Rev. Lett. 36, 56 (1976)
[8] R. T. Tung, Phys. Rev. Lett. 84, 6078 (2000); R. T. Tung, Phys. Rev. B 64, 205310 (2001)
[9] Zhaofu Zhang, et al, ACS Appl. Mater. Interfaces 2015, 7, 5141−5149
[10] Zhaofu Zhang, et al, ACS Appl. Mater. Interfaces 2018, 10, 17419−17426
[11] Zhaofu Zhang et al, Appl. Phys. Express 11 081003 (2018)
[12] S. P. Kowalczyk, et al, Phys. Rev. Lett. 44, 1620 (1980); Su-Huai Wei and Alex Zunger, Appl. Phys. Lett. 72, 2011 (1998); Su-Huai Wei, et al, Appl. Phys. Lett. 94, 212109 (2009)
[13] C. G. Van de Walle and R. M. Martin, Phys. Rev. B 34, 5621 (1986); A. Baldereschi, S. Baroni, and R. Resta, Phys. Rev. Lett. 61, 734 (1988); Alfredo Pasquarello, et al, Appl. Phys. Lett. 107, 211601 (2015)
[14] J. O. McCaldin, T. C. McGill, and C. A. Mead Phys. Rev. Lett. 36, 56 (1976); Kowalczyk, S. P.; Cheung, J. T.; Kraut, E. A.; Grant, R. W. Phys. Rev. Lett. 56, 1605 (1986).
https://m.sciencenet.cn/blog-2686986-1172373.html
ЩЯвЛЦЊЃКвдКѓДђЫуВЛЖЈЦкдкетРяУцаДвЛаЉЬћзг
ЯТвЛЦЊЃКБэУцНчУцНЈФЃвЊЕу/ОбщаЁНс Summary about surface/interface modeling
ШЋВПзїепЕФЦфЫћзюаТВЉЮФ
- • [зЊди]НщЩмЯТздМКЕФзюаТЙЄзїЃКЛњЦїбЇЯАМгЫйЙ§ЖЩН№ЪєЬЊнМЫЋН№ЪєЮЛЕуДпЛЏМСЕФCO2 ЛЙдЗДгІДпЛЏЛюаддЄВт
- • НщЩмЯТздМКЕФзюаТЮФеТAFM: РэТлНвЪОЕЅдзгДпЛЏМСЯѕЫсбЮбЁдёадКЯГЩАБЕФЕчДпЛЏЛњРэ
- • аЁЬИCASTEP(Linux), MS-CASTEP, VASPШэМўЧјБ№
- • БэУцНчУцНЈФЃвЊЕу/ОбщаЁНс Summary about surface/interface modeling
- • вдКѓДђЫуВЛЖЈЦкдкетРяУцаДвЛаЉЬћзг
ШЋВПОЋбЁВЉЮФЕМЖС
ЯрЙиВЉЮФ
- • ЮЊЪВУДЫЕбЇепвЊвдХњХаадЫМЮЌЮЊЦ№Еу
- • [зЊди]ЁОЯВБЈЁПЁЖжЧФмПЦбЇгыММЪѕбЇБЈЁЗ14ЮЛБрЮЏШыбЁ2023ЁАжаЙњИпБЛв§бЇепЁБ
- • ЫяУїшЄНЬЪкШыбЁ2023ФъАЎЫМЮЈЖћИпБЛв§бЇеп
- • [зЊди]ЮваЃдјШйВ§НЬЪкСЌај3ФъШыбЁАЎЫМЮЈЖћЁАжаЙњИпБЛв§бЇепЁБ
- • ЯВбЖЃЁ16ЮЛNMLБрЮЏШыбЁ2023АЎЫМЮЈЖћЁАжаЙњИпБЛв§бЇепЁБАёЕЅ
- • [зЊди]JAS 103УћзЈМвШыбЁАЎЫМЮЈЖћ2023ЁАжаЙњИпБЛв§бЇепЁБАёЕЅ