博文
对"The Hallmarks of Cancer"的批评文章
||||
对"The Hallmarks of Cancer"的批评文章
Hanahan和Weinberg 的Review paper: "The Hallmarks of Cancer"已经是癌症研究的最为影响深远的文章,目前已经有上万次的引用。阅读一下对已经教条化的癌症理论的批评,其实是一个不可多得的学习机会,无疑拓展我们的眼界,免除步教条化的思想的后尘。
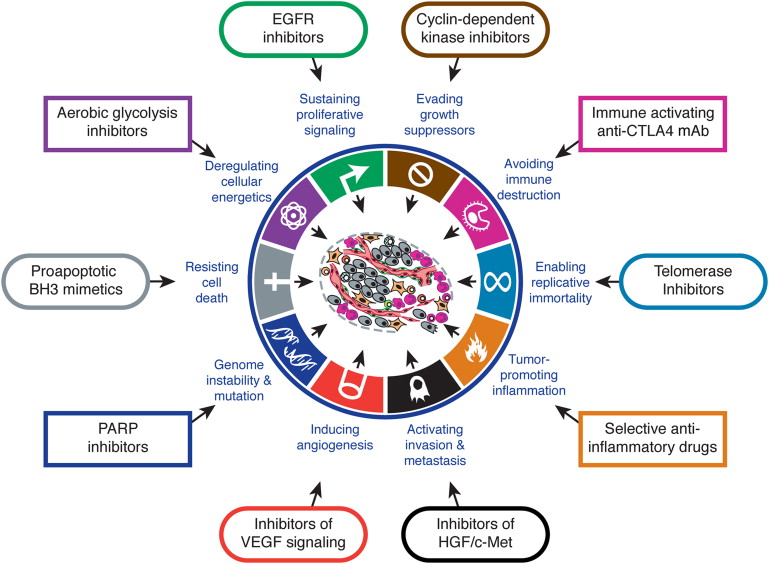
对"The Hallmarks of Cancer"的批评文章
SonnenscheinSoto2013_TheAgingHallMarksofCancer-Critique_JBiosci__frAuth (1).pdf
Wiki;
http://en.wikipedia.org/wiki/The_Hallmarks_of_Cancer

"The Hallmarks of Cancer"[1] is a seminal[2][3] peer-reviewed article published in the journal Cell in January 2000 by US cancer researchers Douglas Hanahan and Robert Weinberg.
The authors believe that the complexity of cancer can be reduced to a small number of underlying principles. The paper argues that all cancers share six common traits ("hallmarks") that govern the transformation of normal cells to cancer (malignant or tumor) cells.
Those hallmarks are: (1) cancer cells stimulate their own growth; (2) they resist inhibitory signals that might otherwise stop their growth; (3) they resist their own programmed cell death (apoptosis); (4) they stimulate the growth of blood vessels to supply nutrients to tumors (angiogenesis); (5) they can multiply forever; and (6) they invade local tissue and spread to distant sites (metastasis).
By November 2010, the paper had been referenced over 15,000 times by other research papers, and was downloaded 20,000 times a year between 2004 and 2007.[4][self-published source?] As of March 2011 it was Cell's most cited article.[2]
In 2011, Weinberg and Hanahan proposed 4 new hallmarks: (1) abnormal metabolic pathways; (2) evading the immune system; (3) chromosome abnormalities and unstable DNA; and (4) inflammation.[5]
Cancer cells have defects in the control mechanisms that govern how often they divide, and in the feedback systems that regulate these control mechanisms (i.e. defects inhomeostasis).
Normal cells grow and divide, but have many controls on that growth. They only grow when stimulated by growth factors. If they are damaged, a molecular brake stops them from dividing until they are repaired. If they can't be repaired, they commit cell suicide (apoptosis). They can only divide a limited number of times. They are part of a tissue structure, and remain where they belong. They need a blood supply to grow.
All these mechanisms must be overcome in order for a cell to develop into a cancer. Each mechanism is controlled by several proteins. A critical proteins must be damaged in each of those mechanisms. These proteins are damaged when the DNA sequence of their genes is damaged through acquired or somatic mutations (mutations that are not inherited but occur after conception). This occurs in a series of steps, which Hanahan and Weinberg call hallmarks.
Self-sufficiency in growth signalsCancer cells do not need stimulation from external signals (in the form of growth factors) to multiply.Normal cells require external growth signals (growth factors) to grow and divide. These signals are transmitted through receptors that pass through the cell membrane. When the growth signals are absent, they stop growing.
Cancer cells can grow and divide without external growth signals. Some cancer cells can generate their own growth signals. For example, glioblastomas can produce their own platelet-derived growth factor (PDGF), and sarcomas can produce their own tumor growth factor α (TGF-α).
Receptors themselves can be overexpressed. For example, the epidermal growth factor receptor (EGF-R/erbB) is overexpressed in stomach, brain and breast cancers, while the HER2/neu receptor is overexpressed in stomach and breast cancer. Or, mutated receptors can send signals without any growth factors at all.
Insensitivity to anti-growth signalsCancer cells are generally resistant to growth-preventing signals from their neighbours.

The growth of normal cells is kept under control by growth inhibitors in the surrounding environment, in the extracellular matrix and on the surfaces of neighboring cells. These inhibitors act on the cell cycle clock, by interrupting cell division (mitosis) in the interphase.
Ultimately, the growth inhibitor signals are funneled through the downstream retinoblastoma protein (pRB), which prevents the inappropriate transition from (G1) to S. If pRB is damaged through a mutation in its gene, or by interference from human papillomavirus, the cell can divide uncontrollably, which can lead to cervical cancer.
Evading apoptosisApoptosis is a form of programmed cell death (cell suicide), the mechanism by which cells are programmed to die in the event they become damaged. Cancer cells characteristically are able to bypass this mechanism.Apoptosis can be triggered by an overexpressed oncogene, and this may be the primary means by which such mutant cells are continually removed. Conversely, cancer cells must overcome apoptosis to progress.
The apoptotic machinery can be divided into sensors, which monitor the cell for abnormal behavior, and effectors, which cause apoptosis.
The sensors include survival signals and their receptors, which monitor the cell for DNA damage, oncogene overexpression, and low oxygen (hypoxia). They monitor survival signals from the cell matrix and neighboring cells.
Sensors include IGF-1/IGF2 and their receptor IGF-1R; and IL-3 and its receptor.
The effectors include FAS ligand and its receptor, and TNF-α and its receptor.
The p53 tumor suppressor protein elicits apoptosis in response to DNA damage, and is a major mechanism of cancer control. In order for cancer to progress, it must overcome p53, and p53 is mutated in >50% of cancers.
Limitless reproductive potentialNon-cancer cells die after a certain number of divisions. Cancer cells escape this limit and are apparently capable of indefinite growth and division (immortality). But those immortal cells have damaged chromosomes, which can become cancerous.Mammalian cells have an intrinsic program, the Hayflick limit, that limits their multiplication to about 60–70 doublings, at which point they reach a stage of senescence.
This limit can be overcome by disabling their pRB and p53 tumor suppressor proteins, which allows them to continue doubling until they reach a stage called crisis, with apoptosis, karyotypic disarray, and the occasional (10−7) emergence of an immortalized cell that can double without limit. Most tumor cells are immortalized.
The counting device for cell doublings is the telomere, which loses DNA at the tips of every chromosome during each cell cycle. Many cancers involve the upregulation of telomerase, the enzyme that maintains telomeres.
Sustained angiogenesisAngiogenesis is the process by which new blood vessels are formed. Cancer cells appear to be able to kickstart this process, ensuring that such cells receive a continual supply of oxygen and other nutrients.Cancer cells initially lack angiogenic ability, limiting their ability to expand. In order to progress, they must develop a blood supply. Angiogenesis is balanced by inducers and inhibitors.
Inducers include vascular endothelial growth factor (VEGF) and acetic and basic fibroblast growth factor (FGF 1/2), which bind to transmembrane tyrosine kinase receptors displayed on endothelial cells. An inhibitor is thrombospondin-1, which binds to CD36. Thrombospondin-1 is regulated by p53, so loss of p53 can allow angiogenesis.
Angiogenesis is involved in the growth of cervix, breast and melanoma tumors.
Anti-VEGF antibodies slowed the growth of tumors in mice. So this and other anti-angiogenesis compounds are under investigation as drugs to treat cancer.
Tissue invasion and metastasisCancer cells can break away from their site or organ of origin to invade surrounding tissue and spread (metastasize) to distant body parts.Primary tumor masses spawn "pioneer cells" that invade adjacent tissues, and may then travel to distant sites, and establish metastases.
The newly formed metastases arise as amalgams of cancer cells and normal supporting cells conscripted from the host tissue.
Metastatic cells must mimic normal cell–cell interactions, through cell–cell adhesion molecules (CAMs) and integrins. N-CAM is normally adhesive, suppressing metastases, but it becomes altered and allows metastases in Wilm's tumor, neuroblastoma, and small cell lung cancer, and its expression is reduced in invasive pancreatic and colorectal cancers.
E-cadherin, which is expressed on epithelial cells, transmits antigrowth signals. E-cadherin is therefore a widely acting suppressor of invasion and metastasis by epithelial cells, which must be overcome by cancer cells to progress.
Integrins display substrate preferences, and changes in integrins are displayed by migrating cells.
Matrix-degrading proteases are also necessary to facilitate invasion into stroma, across blood vessel walls, and through noral epithelial cell layers.
Updates – 2010In his 2010 NCRI conference talk, Hanahan proposed four new hallmarks:
Deregulated metabolismMost cancer cells use abnormal metabolic pathways to generate energy, a fact appreciated since the early twentieth century with the postulation of the Warburg hypothesis,[7] but only now gaining renewed research interest.[8]Evading the immune systemCancer cells appear to be invisible to the body’s immune system.Unstable DNACancer cells generally have severe chromosomal abnormalities, which worsen as the disease progresses.InflammationRecent discoveries have highlighted the role of local chronic inflammation in inducing many types of cancer.CriticismsAn article in Nature Reviews Cancer in 2010 pointed out that five of the 'hallmarks' were also characteristic of benign tumours.[9] The only hallmark of malignant disease was its ability to invade and metastasize.[9]
Notes and references^ Hanahan D, Weinberg RA (January 2000). "The Hallmarks of Cancer". Cell 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931.
^ a b "Scientists Revisit 'Hallmarks of Cancer'". Science Daily. 16 March 2011. Retrieved 2011-04-04.
^ "The Hallmarks of Cancer". Science Interviews. November 2010. Retrieved 2011-04-04.
^ Hallmarks of Cancer, Cancer Research UK Science Update blog, Nov 2010
^ Hanahan, D.; Weinberg, R. A. (2011). "Hallmarks of Cancer: The Next Generation". Cell 144 (5): 646–674.doi:10.1016/j.cell.2011.02.013. PMID 21376230.
^ O. Warburg, K. Posener, E. Negelein: "Ueber den Stoffwechsel der Tumoren" Biochemische Zeitschrift, 152, pp. 319–344, 1924. (German). Reprinted in English in the book On metabolism of tumors by O. Warburg, Publisher: Constable, London, 1930.
^ "Targeting tumour metabolism". Nature Reviews Drug Discovery 9 (7): 503–504. 2010. doi:10.1038/nrd3215.ISSN 1474-1776. PMID 20592733.
^ a b Lazebnik Y (April 2010). "What are the hallmarks of cancer?". Nat. Rev. Cancer 10 (4): 232–3.doi:10.1038/nrc2827. PMID 20355252.
https://m.sciencenet.cn/blog-3468-723923.html
上一篇:达克效应和中国科技落后的原因
下一篇:癌症药物能将癌细胞赶尽杀绝吗?