博文
代谢学人--Science:中暑为啥会死人?
||
代谢学人
Science:中暑为啥会死人?
撰文 | 于柳 徐梓禾 陈俊桐 徐鑫铭 邱瑾
编辑 | 孟美瑶
校对 | 陈俊桐

背景介绍
中暑是一种由热刺激引发的危及生命的疾病,与循环衰竭和多器官功能障碍有关。据估计,如果全球持续变暖,中暑可能会成为全球范围内更重要的死亡原因。临床上,中暑的特征是极端高温、全身炎症反应、循环系统衰竭、出血和凝血功能障碍以及多器官功能障碍,这是由热相关的细胞毒性、炎症和弥散性血管内凝血(DIC)之间复杂的相互作用造成的,而这其中的分子机制仍不明确。
有研究发现,在秀丽隐杆线虫中,热刺激反应通过调节钙网蛋白和钙蛋白酶引起普遍的细胞死亡,而这些蛋白质的消耗则能够限制热应激诱发的线虫死亡。在脊椎动物中,热刺激反应会激活混合谱系激酶样结构域(MLKL)或gasdermin(如gasdermin D,GSDMD)家族蛋白,这些蛋白会通过在细胞质膜或内膜上形成气孔来介导细胞死亡。其中,MLKL能够被受体相互作用蛋白激酶3 (RIPK3)磷酸化而激活,诱导细胞坏死;GSDMD则能够被caspase-1/4,5/11激活,触发细胞焦亡(另一种细胞炎症性坏死形式),其过度激活会导致脓毒症(一种临床上类似于中暑由感染引发的危重疾病)中的DIC和多器官功能障碍。
本文研究了细胞程序性死亡在中暑发病中的可能作用。发现一种Z-核酸传感器,即Z-DNA结合蛋白1 (ZBP1),通过触发RIPK3诱导的MLKL依赖性细胞坏死,以及在较小程度上触发caspase-8(casp8)依赖性细胞死亡,从而介导中暑的病理特征。

敲黑板啦!
1、RIPK3介导热刺激诱导的细胞死亡和中暑特征
2、热刺激通过ZBP1激活RIPK3触发细胞死亡
3、热刺激通过HSF1增加ZBP1的表达
4、热刺激通过RHIM结构域激活ZBP1
5、热刺激促进ZBP1融合蛋白的聚集
研究结果
1. RIPK3介导热刺激诱导的细胞死亡和中暑特征
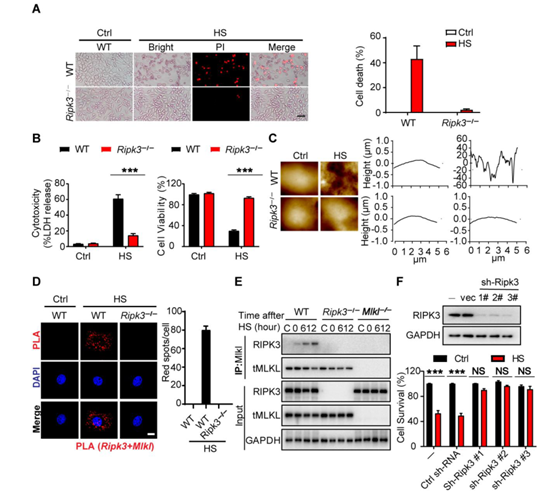
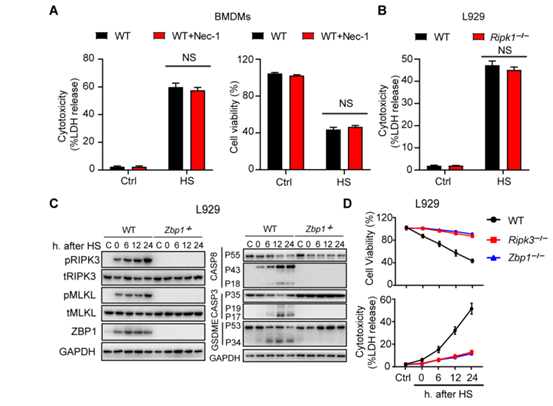
为了研究细胞程序性死亡在中暑发病机制中的作用,作者将Ripk3−/−、Mlkl−/−和 Mlkl−/−Casp8−/−小鼠和相应的野生型(WT)对照小鼠置于温度可控的培养箱中,设置温度条件为39°C,相对湿度为60 ± 5%来诱导中暑。随着时间的推移,在各组织中测量细胞程序性死亡标志物。发现,在2小时内热刺激效应使所有小鼠的核心温度提高到43°C,并且在野生型小鼠中诱发了肝、肺、肠中RIPK3和MLKL的磷酸化,同时伴随着血清中RIPK3浓度的升高(图1、A、B和图S1A)。与此同时,通过蛋白免疫印迹实验,作者发现热刺激还诱导了pro-casp8、pro-casp3、GSDME和GSDMD的激活(图1A和图S1A)。但在Ripk3−/−小鼠中,热刺激诱导的细胞程序性死亡途径的激活作用被显著削弱(图1A和图S1A)。
此外,作者发现热刺激还会增加血清中乳酸和促炎细胞因子(其中包括白细胞介素-1 (IL-1)、IL-6和肿瘤坏死因子(TNF)等)的浓度,可极大的引发小鼠体内电解质紊乱(小编注:尽管没有明确的文章说明乳酸和炎症因子直接促进电解质紊乱,但根据电解质紊乱的诱因之一可以推测可能是血清中不溶物质和可溶物质增加时,可以使单位体积的水含量减少,血钠浓度降低,从而引发电解质紊乱。具体电解质紊乱的指标可能是通过实验数据检测体液中Na+、K+、Ca2+、Mg2+、Cl−、HCO3−发现的),而这些效应在Ripk3−/−小鼠中均被削弱(图1C和图S1、B和C)。同时,热刺激也可诱导血管内凝血酶生成、血小板聚集、纤维蛋白沉积、微循环闭塞和循环DIC标志物增加(小编注:DIC可加重危重病人的多器官损伤以及提高致死率。本文章作者此前研究过危重症的发病机制,其中脓毒症与热射病均表现为全身炎症反应、DIC和多脏器衰竭。作者前期研究发现脓毒症通过HMGB1-caspase-11途径诱发多种程序性细胞死亡进而诱导DIC,而本文发现热射病导致的热应激与脓毒症相同,也是通过诱发过度的细胞程序性死亡进而诱导DIC,与脓毒症不同的是,热应激诱发细胞程序性死亡的途径为RIPK3途径),而这些效应都可被Ripk3缺乏所减弱(图1、D、E和图S1、D、E)。与这些临床观察结果相一致,RIPK3缺失可减轻热刺激诱发的肝、肺和肠损伤;预防肾功能和肺功能障碍;以及降低致死率(图1 F 、G, 和图S1、F-J)。因此,结果表明RIPK3参与并介导了中暑的病理性特征。
先前研究表明,RIPK3以激酶依赖的方式通过MLKL介导细胞坏死,并通过casp8以激酶非依赖形式来诱导细胞凋亡和坏死。为了研究RIPK3在热刺激中的辅助功能,作者构建了Ripk3∆/∆小鼠,这些小鼠可以表达由RIPK3激酶结构域中四个氨基酸(QWDF)缺失所致的催化活性不高的的RIPK3。检测发现,RIPK3激酶活性丧失抑制了热刺激诱导的MLKL磷酸化作用,但不影响pro-casp8、pro-casp3和GSDME的裂解活化(图1H和图S2A)。热刺激后RIPK1激酶结构域的失活并不影响死亡标志物的水平(图1H和图S2A)。当casp8和MLKL同时缺失时,却能够阻断pro-casp3和GSMDE的裂解活化,并减少GSDMD的活化,抑制细胞死亡(图1A)。
接下来,作者检测了RIPK3激酶依赖和激酶不依赖途径在热刺激相关的致死性中的作用。敲除RIPK3可以几乎获得全部保护,而只是丧失RIPK3的激酶活性可以拯救70%致死热刺激后的小鼠(图1I)。与这些观察结果一致,敲除MLKL后可减轻器官损伤,拯救了75%的小鼠,而同时敲除MLKL和casp8则可使所有热刺激下的小鼠存活(图1I)。GSDMD的缺乏则提供了相对次要的保护(图S2B)。实验结果显示,与WT小鼠一样,热刺激使Ripk3−/−和Mlkl−/−Casp8−/−小鼠的核心温度均升高到43°C(图S2C),从而排除了体温降低促进小鼠存活率升高的可能性。因此,这些发现支持了热刺激中RIPK3依赖的细胞死亡的重要作用。
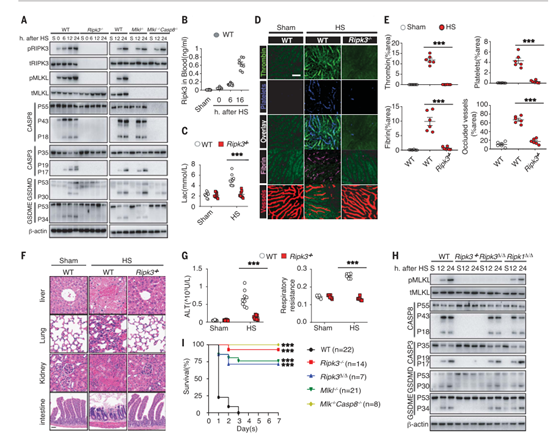
图1. RIPK3介导热刺激诱导的细胞死亡和中暑特征
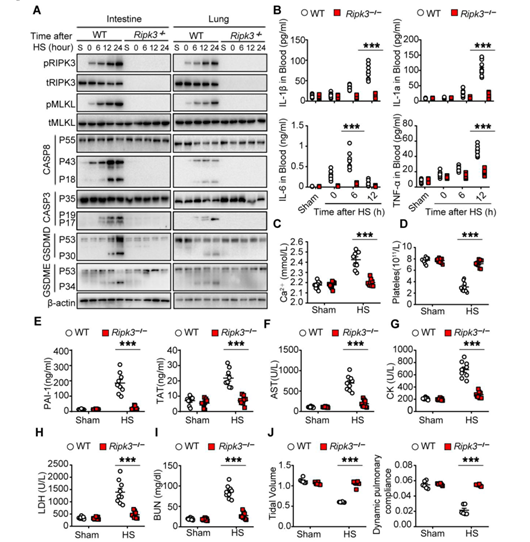
附图1
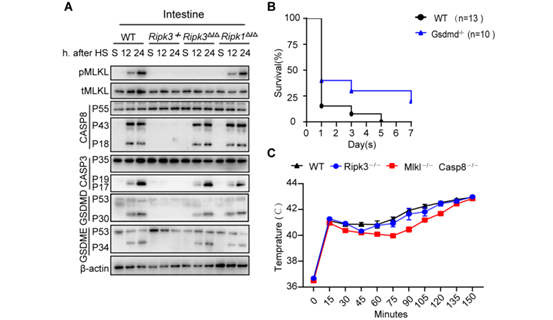
附图2
拓展阅读
弥散性血管内凝血(DIC)
弥散性血管内凝血(disseminated intravascular coagulation,DIC)不是一种独立的疾病,而是许多疾病在进展过程中产生凝血功能障碍的最终共同途径,是一种临床病理综合征。
DIC分为急性DIC和慢性DIC。急性DIC是一种消耗性凝血病状态。在这种状态下,一方面,过量凝血酶抑制了血浆中存在的天然抗凝剂,血液内凝血机制被弥散性激活,促发小血管内广泛纤维蛋白沉着,造成多器官功能障碍。另一方面,凝血酶的促凝作用也促进抗凝血酶的生成,形成更多的纤维蛋白凝块,大量的促凝物质入血,凝血作用导致微循环中形成广泛的微血栓,继而因凝血因子和血小板被大量消耗,引起继发性纤溶亢进,这最终导致纤维蛋白溶解,纤维蛋白血栓被纤溶酶分解,随后释放纤维蛋白降解产物(FDPs)。在血管内时,这些FDP可以通过干扰GPIIbIIIa纤维蛋白原受体来抑制纤维蛋白聚合和血小板聚集。因此,FDP与血小板、纤维蛋白原和凝血因子的消耗共同导致急性DIC中第二种常见症状:出血。血栓和出血两种矛盾的表现在DIC疾病发展过程中同时存在,并构成特有临床表现。
休克也通常发生在急性DIC中,可导致脾脏和肝脏的网状内皮系统巨噬细胞无法清除组织因子、活化凝血因子和FDP,从而使DIC永久化。当身体长时间暴露于少量凝血酶时(即恶性肿瘤、宫内胎儿死亡、血管炎、动脉瘤、血管瘤和大面积愈合等),可能会发生慢性DIC。虽然凝血因子和血小板被消耗,但不如急性DIC时的消耗大,身体能够通过增加凝血因子、血小板、抗凝血酶和抗血浆的生成进行部分补偿。此外,肝仍能有效清除FDP。因此,血栓形成通常是慢性DIC出血的主要原因,在这种情况下通常不存在休克。
在本文中,作者发现中暑伴随着DIC症状的发生,并且DIC可加重危重病人的多器官损伤以及提高致死率。
参考文献:
[1] Boral BM et al. Am J Clin Pathol. 2016.
2.热刺激在体外通过激活RIPK3触发细胞死亡
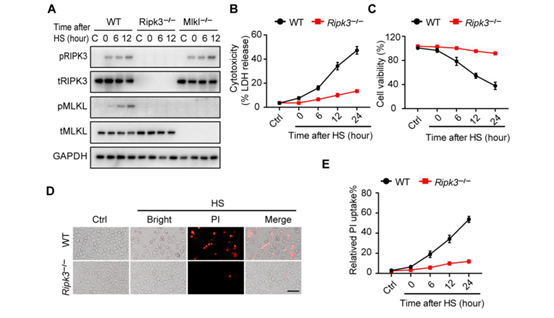
作者为了研究热刺激是否能够单独激活RIPK3依赖的细胞死亡,将培养的L929小鼠成纤维细胞暴露在43°C下2小时或42°C下6小时,发现可在2小时内诱导RIPK3和MLKL磷酸化,并在热刺激暴露6小时后诱导pro-casp8、pro-casp3和GSDME裂解(图2A)。当使用CRISPR-CAS9系统敲除Ripk3后,则可以阻断热刺激诱导的RIPK3和MLKL磷酸化以及pro-casp8、procasp3和GSDME裂解(图2A)。同时,敲除Ripk3后,细胞乳酸脱氢酶(LDH)的释放、细胞膜完整性的丧失和细胞膜通透性的增加等不良反应均被显著降低,说明热刺激介导的细胞死亡被抑制(图2、B和C和图3 A-C)。热刺激促进了RIPK3和MLKL在WT小鼠中的相互作用,而在Ripk3缺失的L929细胞中并没有这种效果(图S3,D和E)。与此同时,作者用shRNA沉默RIPK3的表达,发现可以抑制热刺激诱发的细胞死亡(图S3 F);而回复RIPK3的表达后,Ripk3缺陷的L929细胞又出现了热刺激时细胞死亡的现象(图2 D)。此外,作者对WT或Ripk3−/−小鼠的骨髓源性巨噬细胞(BMDMs)和腹膜巨噬细胞(PMs)进行观察,也发现了类似的现象(图2,E和F,图S4, A至E)。尽管Mlkl缺陷可以在12小时内保护细胞免受热刺激诱导的坏死,但在暴露24小时后Mlkl−/−细胞仍会出现死亡,而Ripk3−/−BMDMs则没有类似的现象,细胞坏死被完全抑制(图2E)。有研究报道,由于RIPK3可以作为支架蛋白(非激酶活性)来介导casp8,引发细胞死亡(小编注:K. Newton et al., 在2014年的Science研究中发现, RIPK3和RIPK1通过它们的RIP同型相互作用基序(RHIMs)相互作用;RIPK1和FADD可以通过其死亡域相互作用,FADD和caspase-8通过其死亡效应域相互作用,最终促进caspase-8裂解激活,如下图),因此作者认为热刺激可能通过RIPK3来诱导MLKL和casp8依赖的细胞死亡。
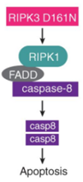
RIPK3作为支架蛋白介导casp8激活
(K. Newton et al., Science 343, 1357–1360 (2014).)
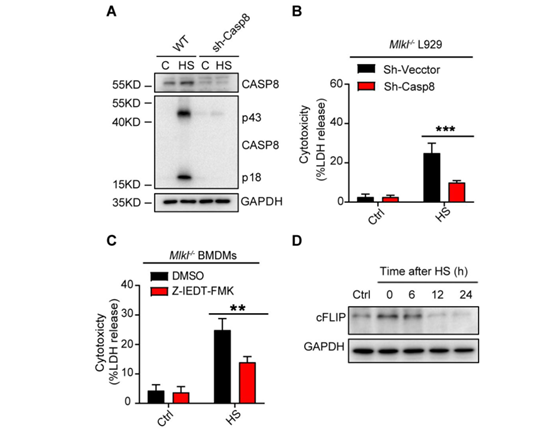
为了验证这一观点,作者将WT、Ripk3∆/∆、Mlkl−/−、和 Mlkl−/−&Casp8−/−BMDMs暴露于热刺激环境中。研究发现,RIPK3激酶活性的丧失阻止了MLKL磷酸化,但没有阻止casp8、casp3和GSDME的裂解活化(图2F)。而MLKL和casp8的缺失几乎完全阻止了casp3和GSDME的裂解活化(图2F)。在热刺激后6小时,RIPK3的激酶活性丧失或Mlkl缺失可以阻止LDH释放和细胞死亡,但来自Ripk3∆/∆或Mlkl−/−小鼠的BMDMs在热刺激暴露24小时后会释放LDH并发生细胞死亡(图2E)。然而,通过敲除Ripk3或同时敲除Mlkl和casp8,可以持续的抑制LDH释放和细胞死亡(图2E)。利用shRNA沉默casp8的表达或用Z-IEDT-FMK对casp8进行药物抑制,可以减少热刺激诱导的Mlkl缺失引起的L929细胞的死亡(图S5, A-C)。此外,热刺激短时间内(12小时内)增加了细胞FLICE-样抑制蛋白(cFLIP)的表达(图S5D),而cFLIP可以抑制casp8激活和及其相关的细胞死亡,说明在热刺激前期主要是RIPK3激酶激活的MLKL来发挥作用,后期casp8激活后也可以促进细胞死亡。因此,热刺激通过RIPK3依赖的MLKL和casp8激活途径共同来触发细胞死亡。
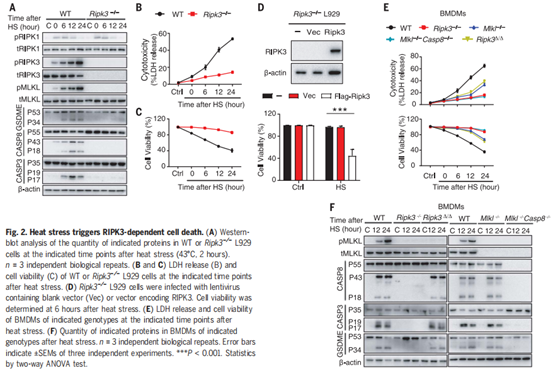
图2. 热刺激触发Ripk3依赖的细胞死亡
附图3
附图4
附图5
拓展阅读
Caspase的好伙伴:MLKL与GSDMD
caspase家族即半胱氨酸蛋白酶,在细胞凋亡过程中发挥重要作用。目前有研究发现本文所涉及到的caspase8(促凋亡Caspases)可以介导细胞凋亡途径之间的可塑性,caspase-8可以抑制由RIPK3激酶和MLKL介导的细胞坏死,缺乏caspase-8的小鼠表现出MLKL依赖的胚胎致死性;而caspase家族(促炎Caspases)与GSDMD之间的关系表现为被激活的caspase切割GSDMD蛋白,释放出其N端结构域,该结构域结合膜脂并在细胞膜上打孔,导致细胞渗透压的变化,进而发生胀大直至细胞膜破裂,细胞发生焦亡,抵抗感染作用。例如,caspase-1是静止细胞中的一种休眠酶。在感染或细胞稳态被选择性破坏时,炎症小体在细胞质中组装,招募和激活caspase-1。caspase-1将GSDMD切割形成两个片段,N端片段聚合并插入质膜,可形成内径10-20nm的孔隙。
参考文献:
[1]Ding, J. et al.Nature 535, 111-116, doi:10.1038/nature18590 (2016).
拓展阅读
乳酸脱氢酶(LDH)试验与组织损伤
乳酸脱氢酶 (LDH) 是在大多数生物体内发现的一种酶,可以将糖酵解的终产物丙酮酸转化为乳酸。在转化过程中,消耗一个单位的能量,NADH释放氢产生NAD+,以继续糖酵解。乳酸脱氢酶由 4 个不同的亚基组成,这 4 个亚基可以以不同的形式出现,并由不同的基因编码。在人体内,乳酸脱氢酶有 5 种异构体,不同的构象存在于不同的身体组织中,这常常可以帮助医者确定乳酸脱氢酶的来源。例如,LDH-1(乳酸脱氢酶-1)存在于心脏、血细胞和脑细胞中,而LDH-3 仅存在于肺部。
乳酸脱氢酶试验可用于检测组织损伤。此外,由于酶会从受损的组织中释放出来并且不同类型的酶存在差异,医生可以使用乳酸脱氢酶测试来确定身体损伤的部位和程度。例如,受损的心脏组织会释放乳酸脱氢酶,于是最近心脏病发作的人血液中的乳酸脱氢酶水平会升高。医生可以测试这种酶,并确定它确实是 LDH-1,即在心脏组织中发现的酶形式。根据血液或脊髓液中监测的水平,可以估计出心脏组织受损范围以及受损时长。乳酸脱氢酶测试还可用于寻找许多其他疾病,通常监测或诊断内部组织损伤、监测引起损伤的状况或评估某些癌症的治疗。该测试通常与许多其他指标结合使用,血液或脊髓液中高水平的乳酸脱氢酶通常表明组织损伤。
本文中,作者就通过乳酸脱氢酶(LDH)的监测来作为细胞死亡的一个重要指示。
乳酸脱氢酶-M4(肌肉)
参考文献:
[1] Biologydictionary.net Editors. Lactate Dehydrogenase. Biology Dictionary. 2018.
3.热刺激通过ZBP1激活RIPK3
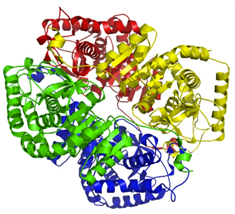
先前研究发现,RIPK1具有包含干扰素-β诱导契合的Toll/白介素-1受体结构域(TRIF),它和ZBP1都是含有RIP同型相互作用基序(RHIM)的蛋白,ZBP1可以与RIPK1发生相互作用并激活RIPK3。因此,为了检测与RIPK3结合的蛋白质是否与热刺激诱导细胞死亡相关,作者检测TRIF缺失、RIPK1激酶结构域突变(RIPK1∆/∆)或通过necrostain1抑制RIPK1表达对细胞死亡的影响,发现均不影响热刺激诱导的BMDMs细胞死亡(图3A和图S6, A和B)。在L929细胞中,RIPK1缺失也不显著抑制热刺激诱导的细胞死亡(图S6A)。然而,ZBP1的缺失却能够抑制BMDMs细胞中RIPK3和MLKL的磷酸化,以及casp8、casp3和GSDME的裂解活化;热刺激后细胞死亡(图3,A和B)。同样,这些发现在L929细胞中也得到了证实(图S6, C和D)。恢复ZBP1表达能够恢复热刺激引发细胞死亡的效果(图3C)。与此同时,作者发现在结肠癌细胞系HT-29中表达RIPK3和RIPK1,但不表达ZBP1时,不会发生热刺激诱导的细胞死亡(图3 D)。在HT-29细胞中外源性表达人源ZBP1后则对热刺激诱导的细胞死亡非常敏感(图3D)。此外,检测发现热刺激能够引发ZBP1和RIPK3之间的相互作用(图3,E和F)。总之,这些数据表明,热刺激通过ZBP1来激活RIPK3。
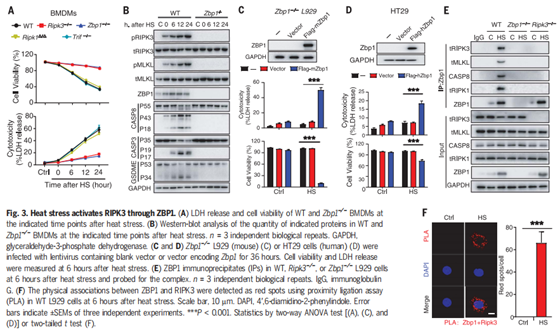
图3. 热刺激通过ZBP1激活RIPK3
附图6
拓展阅读
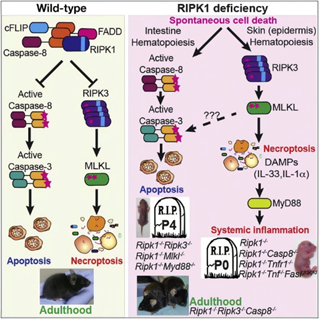
RIPK1与细胞死亡
细胞凋亡(apoptosis)和程序性细胞坏死(necroptosis)是两种不同机制的细胞死亡过程。凋亡是由半胱氨酸酶(caspases)介导的,而当缺乏细胞凋亡的条件时,受体相互作用蛋白激酶1 (Receptor-interacting protein kinase 1, RIPK1)及其下游的RIPK3和MLKL则会激活细胞程序性坏死通路。大量的研究证实RIPK1 是细胞凋亡和坏死以及炎症通路中的一个关键调节因子。因此,RIPK1逐渐成为神经退行性疾病、自身免疫性疾病和炎症等多种疾病治疗的有效靶点之一。
RIPK1是细胞死亡和炎症的关键介质,并且是NF-kB信号转导与死亡反应的细胞决定因素的主要调节剂。RIP3(RIPK3)是丝氨酸/苏氨酸蛋白激酶,可激活坏死和凋亡这两种细胞死亡形式。激活的RIPK3形成一个诱导坏死的复合物,并介导MLKL的磷酸化,促进MLKL定位到质膜并执行程序性坏死,其特征是钙内流和质膜损伤。
现已确定的Caspase家族的蛋白酶至少有11种,并且它们中的大多数都可以直接参与凋亡信号的转导。Caspase8属于半胱氨酸蛋白酶家族,可介导细胞凋亡。以往研究发现,caspase8对于小鼠胚胎发育很重要,caspase8敲除的小鼠会由于过度激活的细胞坏死而引起胚胎致死。RIPK1和Caspase-8在细胞死亡和炎症中独立地确保染色体稳定性。近年来的研究也发现,caspase8可以通过切割RIPK1蛋白从而限制凋亡和坏死信号的过度激活。除此之外,caspase8同时具有诱导和抑制细胞死亡的功能:它可以通过死亡受体来诱导细胞凋亡,同时,也可以通过激酶RIPK3和MLKL来抑制细胞坏死。
程序性细胞坏死可以被TNF家族成员、CD95 (Fas),Toll 样受体3/4 (TLR3/4) 和干扰素 (IFNs) 激活。TNFα与TNF受体1 (TNFR1) 结合,形成由TNFR1相关的死亡结构域蛋白 (TRADD),TNF受体2 (TRAF2),细胞内凋亡蛋白质抑制蛋白1/2 (cIAP1/2) 和RIPK1组成的膜信号复合物I (Complex I)。复合物I能够导致NF-κB通路的激活。在这种情况下,RIPK1被泛素化,从而促进细胞存活。如果抑制cIAP1/2,从而阻断RIPK1泛素化,复合物I将被释放并募集Fas相关的死亡结构域蛋白 (FADD),进而形成复合物II (Complex II)。复合物II可以介导caspases 8的激活,进而激活凋亡信号通路。如果缺乏caspases 8,RIPK1、RIPK3和MLKL将形成坏死体,同时,细胞死亡信号将从凋亡切换到坏死。RIPK3磷酸化坏死体中的MLKL,促使其发生齐聚反应,并定位于细胞膜,导致细胞膜爆裂,促发细胞死亡。
细胞发生凋亡还是坏死,造成的后果大为不同。例如,发生凋亡的细胞形态较为完整、胞浆收缩、细胞核凝集,表现为细胞逐渐变小并固缩成核状,细胞分裂成凋亡小体,细胞器较为完整,不会引起炎症反应。而发生坏死的细胞的细胞膜完整性丧失,细胞和线粒体胀大,最终细胞成碎片状,周围伴有严重的炎症反应。坏死性凋亡则是受到程序性控制的细胞坏死,坏死性凋亡的特征在于细胞体积增大,细胞器肿胀,细胞膜穿孔,之后是细胞崩解,释放内容物,引发周围炎症反应。
参考文献:
[1] Rickard JA1, et al.Cell. 2014 May 7. pii:S0092-8674(14)00537-6.
4.ZBP1介导中暑的病理特征
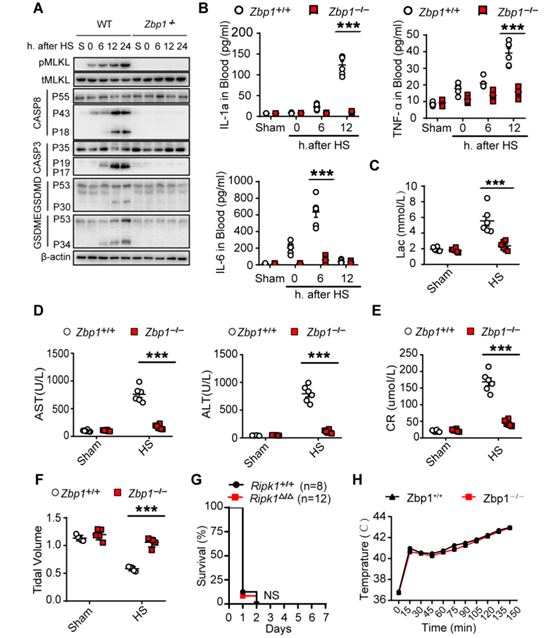
为了确定ZBP1是否通过激活RIPK3并介导热应激后的中暑反应,作者通过将Zbp1−/−小鼠及其WT同窝小鼠暴露于热刺激环境来研究其作用。结果显示,ZBP1缺失阻断了热刺激诱导的RIPK3和MLKL磷酸化以及Casp8、Casp3、GSDMD和GSDME在肝和肠中的裂解活化(图4A和图S7A)。说明ZBP1缺失可预防热刺激诱导的DIC、全身炎症反应、循环衰竭、多器官损伤和致命性细胞坏死特征性反应。在RIPK3缺失小鼠中也观察到了同样的情况(图4,B至F和图S7, B至F)(小编注:前文提到Ripk3∆/∆小鼠热应激39℃ 7天以上仍能保持75%的存活率,而Ripk3−/−小鼠是可以获得将近87.5%以上的存活率,Zbp1−/−则是39℃ 7天87.5%的存活率)。相比之下,TRIF缺乏或RIPK1激酶结构域突变不能保护小鼠免受致命的热刺激的伤害(图4G和图S7G)。与野生型小鼠一样,热刺激使Zbp1−/−小鼠的核心温度升高到43°C(图S7H),也同样排除了Zbp1−/−小鼠存活率升高可能是体温降低的结果这一可能性。这些数据表明,ZBP1介导热刺激诱导的RIPK3激活和中暑的病理特征。
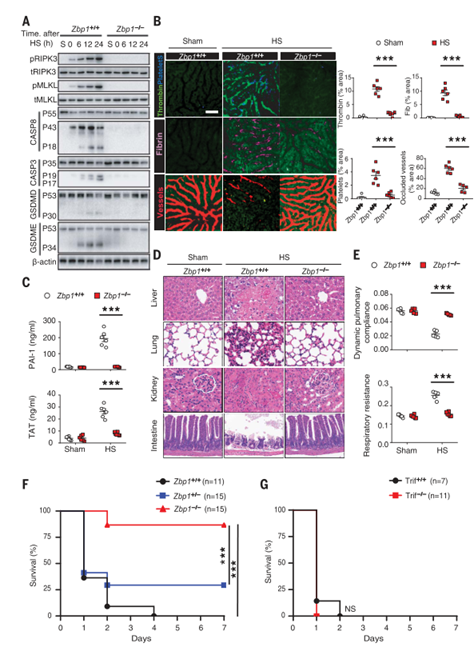
图4. ZBP1介导热刺激诱导的细胞死亡和中暑特征
附图7
5.热刺激通过HSF1增加ZBP1的表达
ZBP1是一种干扰素诱导因子。检测发现,热刺激以类似于诱导表达I型干扰素的方式上调L929细胞和小鼠巨噬细胞中ZBP1的表达(图5,A到D,图S8, A和B)。此外,热刺激还刺激了肺、肝、肾和肠中ZBP1的转录(图5E和图S8C)。作者采用生物信息学工具Jaspar分析了ZBP1的启动子区域,并确定了被热刺激激活的热休克转录因子1 (HSF1)的预测结合位点(图5F),该位点的结合在热刺激下被启动。当缺失HSF1的预测结合位点后,可以阻止热应激后ZBP1的转录激活(图5F)。相反地,热胁迫可增强ZBP1启动子中HSF1结合位点的激活和占用(图5G和图S8, D到F)。HSF1的缺失抑制了热刺激诱导的ZBP1表达增加和细胞死亡(图5H,图S8G,图S9A)。在热刺激反应中,HSF1调节热休克蛋白(HSPs)的表达(图S9B)。有研究证明,HSP90通过促进MLKL的寡聚和激活促进了TNF诱导的坏死。然而,作者发现HSP90的缺乏并不影响热刺激诱导的RIPK3或MLKL的磷酸化和细胞死亡(图S9, C和D)。因此,HSF1介导的ZBP1转录表达在细胞死亡中占有主要的作用。总之,这些数据证实热刺激通过促进HSF1与ZBP1启动子的结合来促进其转录表达。
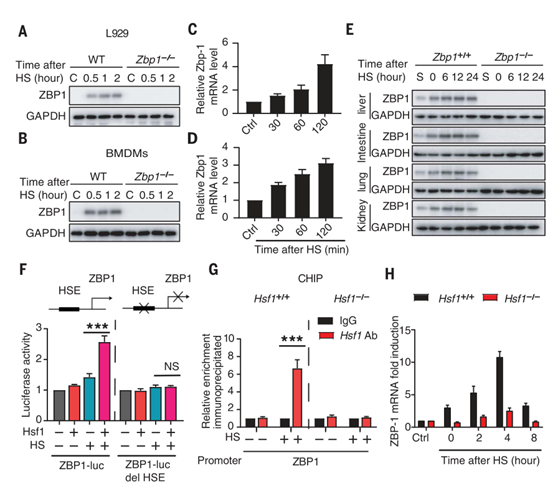
图5. 热刺激通过HSF1增加ZBP1的表达
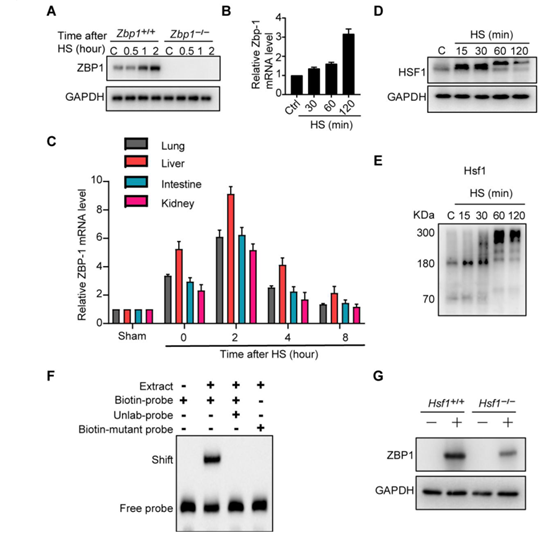
附图8
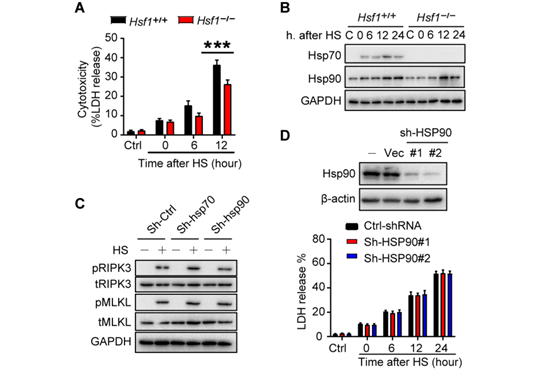
附图9
拓展阅读
热休克蛋白90(HSP90)与细胞死亡
HSP90是一种分子伴侣,能调节相关蛋白质的结构和功能,这些蛋白质与HSP90及其辅伴侣CDC37形成复合物。HSP90功能的丧失可能通过泛素-蛋白酶体途径而导致相关蛋白的不稳定和降解,如RIP1。对HSP90功能的抑制破坏了HSP90和RIP1之间的联系,并导致RIP1的降解。此外,HSP90活性的丧失阻止了TNF诱导的RIP1依赖性NF-κB激活和坏死,并使细胞对TNF诱导凋亡敏感。因此,HSP90是一种伴侣蛋白,在坏死性凋亡途径中维持RIP1的稳定性和功能。
RIP3是调节坏死性凋亡的关键激酶,而HSP90-CDC37复合物在RIP3激活中起到了重要作用。HSP90的抑制和CDC37的敲除都阻止RIP1-RIP3坏死体的形成、RIP3的磷酸化和坏死性凋亡。并且,HSP90活性对于MLKL组装成寡聚体以及这些寡聚物转移到膜上至关重要,通过直接调节MLKL功能来调节坏死性凋亡。
HSP热休克蛋白参与了细胞内的多种生理生化过程,是决定细胞命运的重要蛋白。HSP热休克蛋白对物理、化学和环境胁迫反应迅速且无处不在,共同作用来调节细胞的增殖、生存和死亡。通常认为热休克蛋白(HSP)可以通过阻止蛋白质的变性以及促使变性蛋白复性,保护胁迫条件下的细胞少受损伤。 近年来的研究表明,热休克蛋白家族的重要成员(如HSP90、HSP70、HSP27等)在caspase依赖型细胞凋亡的调节过程中扮演了十分复杂的角色,其参与并调控了细胞凋亡的各个事件,发挥了抗细胞凋亡或促细胞凋亡的作用。一方面,由于热激蛋白对细胞的保护功能,它们抑制细胞凋亡;另一方面,它们又作为一种关键的信号蛋白分子伴侣而直接地促进了细胞凋亡,其中就包括了HSP90。HSP90可协助来自原生质膜的细胞凋亡信号的转导,促进死亡结构域激酶以及受体相关蛋白的活化,从而使细胞对TNF诱导的细胞凋亡十分敏感。
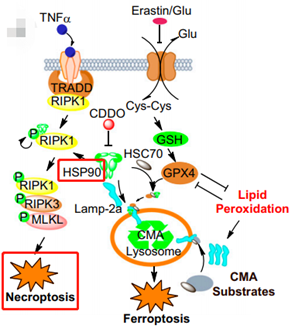
HSP90调节坏死性凋亡
参考文献:
[1]Yang CK et al. Cell Death Dis. 2016
[2]Wu Z, et al. Proc Natl Acad Sci U S A. 2019 Feb 19;116(8):2996-3005.
[3] 摘自《生命科学》:热休克蛋白HSP对细胞凋亡的调节作用
6.Z -核酸感应对于热刺激诱导的ZBP1的激活没有影响
由于只有ZBP1的表达不足以引发细胞死亡(图3,C和D),所以作者进一步研究了热刺激促进ZBP1激活的机制。有研究报道,在发育、病毒感染和其他疾病中,ZBP1被病毒衍生或内源性Z核酸通过其Zα结构域所激活。因此,为了确定Z核酸对ZBP1的影响,作者构建表达完整ZBP1和ZBP1突变体的转基因L929细胞,这些突变体要么缺少Zα、Zα1或Zα2结构域(∆Zα、∆Zα1或∆Zα2),要么包含抑制z -核酸传感的Zα2结构域(Zα2 mut)内的点突变(图6A)。然而,在这些表达ZBP1突变体的L929细胞中,热刺激仍然触发了ZBP1-RIPK3的相互作用,RIPK3和MLKL的磷酸化,以及细胞死亡(图6, B到E)。有研究表明,ZBP1包含一个C端结构域和一个RHIM结构域。检测发现,热刺激诱导的ZBP1-RIPK3相互作用、RIPK3和MLKL磷酸化以及细胞死亡都离不开RHIM而不是C端结构域(图6,B到E)。因此,热刺激通过其RHIM结构域激活ZBP1,而不依赖于Z-核酸的感知。。
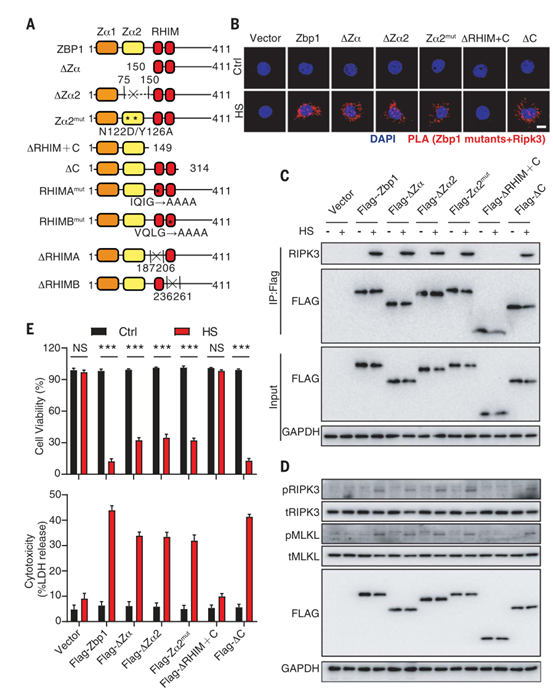
图6. Z -核酸感应对于热刺激诱导的ZBP1的激活没有影响
拓展阅读
ZBP1激活与Zα结构域
Z-DNA结合蛋白1(ZBP1),也被称为IFN调节因子的DNA依赖激活因子,是一种先天的核酸传感器,通过协调细胞死亡和炎症,在调节宿主防御和炎症疾病中发挥作用。ZBP1包含一个RIP同型相互作用基序(RHIM),通过它与其他含有RHIM的蛋白RIPK1和RIPK3相互作用。在含有RHIM的蛋白中,ZBP1的不同之处在于,它还包含核酸感应Zα结构域。与其他核酸传感器不同,ZBP1的Zα结构域在Z-构象中对双链核酸表现出较高的亲和力。右旋DNA(B型)和RNA(A型)在构象上是不同的。然而,采用Z-构象的左旋DNA和RNA表现出相似的结构特征。因此,ZBP1的Zα结构域在体外对Z-DNA和Z-RNA都表现出亲和力。病毒基因组在侵入细胞后会产生Z-结构,Z-DNA或Z-RNA结构是由于病毒复制过程中负超螺旋诱导的扭转压力而产生的,因此ZBP1一旦在细胞核中被激活,就会响应由内而外(即细胞核到细胞质)的细胞死亡信号传导。
最初的研究提出,ZBP1作为一个B-DNA传感器来启动I型干扰素反应。ZBP1还被证明与RIPK3复合物介导M45突变的小鼠巨细胞病毒(DNA病毒)诱导的坏死性凋亡。寻找上游NLRP3炎症小体调节因子的独立研究发现,ZBP1还是甲型流感病毒(IAV,一种RNA病毒)的先天免疫传感器,可触发焦亡、凋亡和坏死性凋亡的执行。ZBP1的Zα结构域在感知RNA病毒感染方面起到重要作用,但目前对于病毒或内源性RNA在Z构象中的生物学意义仍缺乏深入了解。本篇文献中,作者通过突变ZBP1的Zα结构域证实热刺激并不依赖于z -核酸的感知来激活ZBP1。
参考文献:
[1] Kesavardhana et al. J Exp Med. 2020.
7.热刺激促进ZBP1融合蛋白的聚集
为了进一步研究热刺激是如何激活ZBP1的,作者用表达绿色荧光蛋白(GFP)标记的ZBP1的质粒转染HEK 293T细胞。将细胞暴露于43°C下时,发现ZBP1-GFP结合的点状物聚集在胞浆内(图S10A)。检测发现,Zα结构域对热应激诱导的ZBP1-GFP聚集起重要作用(图S10A)。而热刺激条件下ZBP1聚集不受Zα2结构域缺失或Zα2结构域点突变的影响(图S10A)。同时,非还原条件下的免疫印迹实验。结果显示,热刺激诱导内源性ZBP1的聚集(图S10B)。作者利用表达Flag-或myc标记的ZBP1或ZBP突变体的细胞也证实细胞暴露于热刺激增加了ZBP1的聚集(图S10, C和D)。以上发现证明Zα结构域、C端结构域和Z -核酸传感对于ZBP1聚集和细胞死亡是不可或缺的(图S10, C到G)。
先前研究报道,ZBP1包含两个RHIM域,称为RHIM- A和RHIM- B。检测发现,RHIM- A区域内的点突变可以阻止热刺激引发的ZBP1融合蛋白聚合(图S10, A到F)。当用WT ZBP1和RHIM-B缺失或突变的ZBP1重组Zbp1−/− L929细胞,而不是ZBP1 Zα结构域突变和RHIM-A缺失或突变,则能够诱发热刺激后ZBP1融合蛋白的聚合、MLKL的磷酸化,casp8的裂解活化以及细胞死亡(图S10, E到G)。结果显示,ZBP1融合蛋白的聚合最早发生在热刺激的30分钟以后,随后是RIPK3招募、MLKL磷酸化和casp8的裂解(图S10F),这些效应都能被RHIM- A域的缺失或点突变所阻断(图S10F)。此外,作者用HBD二聚体4-羟基他莫西芬(4-OHT)处理表达激素结合结构域的G521R突变体(HBD*)融合ZBP1蛋白的L929细胞(小编注:作者在激素结合结构域(G521R)突变的L929细胞(HBD*)中表达ZBP1的融合蛋白,接着又用HBD二聚体4-羟基塔米芬(4-OHT)处理这些细胞,由于融合蛋白可与4-OHT相互结合并聚集,间接的将HBD-ZBP1聚集(图S10H),发现也可以触发细胞死亡(图S10I))。因此,结果显示热刺激可能是通过加强了ZBP1聚集来促进细胞死亡过程的。
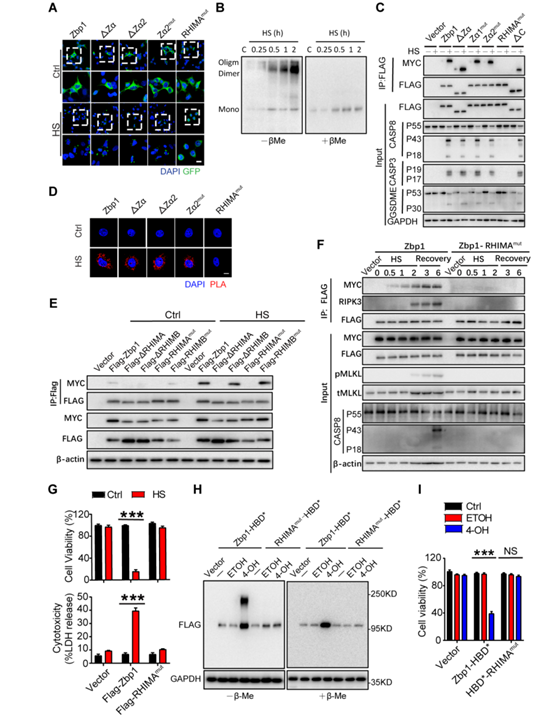
附图10
总结
本研究确定了ZBP1通过Ripk3依赖的细胞死亡促进中暑病理特征的作用。本文介绍的Z-DNA结合蛋白1(ZBP1)是一种Z-核酸受体,它通过触发受体相互作用蛋白激酶3(RIPK3)依赖的细胞死亡来介导中暑。热刺激通过促进热休克转录因子1(HSF1)在ZBP1启动子附近的结合来提高ZBP1的转录表达,并通过一种独立于核酸感应的机制来激活ZBP1(即热刺激通过其RHIM结构域激活ZBP1),进而促进其蛋白聚集来促进细胞死亡。
此外,敲除ZBP1、RIPK3或同时敲除混合谱系激酶样结构域(MLKL)与caspase-8可改善热激诱导的循环衰竭、器官损伤和致死率。因此,ZBP1可能具有调节机体对热激反应的新功能。
原文链接:https://www.science.org/doi/10.1126/science.abg5251
关注微信公众号代谢学人
了解更多代谢前沿资讯

https://m.sciencenet.cn/blog-3483272-1351799.html
上一篇:代谢学人--Nature:凋亡的脂肪细胞也要发挥余温!
下一篇:代谢学人-Cell&Science&Nature近期代谢精选