博文
214无血清纯化Vero狂犬病疫苗在健康成人中的安全性和免疫原性:一项随机II期暴露前预防研究
||
无血清纯化Vero狂犬病疫苗在健康成人中的安全性和免疫原性:一项随机II期暴露前预防研究
突出
三剂PVRV-NG暴露前方案在免疫原性上不劣于HDCV。
98.3%的PVRV NG受试者的中和抗体滴度≥ 0.5 IU/mL。
用PVRV-NG进行的初级暴露前免疫有效地引发了对加强免疫的强烈免疫反应。
在健康成人中,PVRV-NG具有可接受的安全性,耐受性良好。
摘要
一种无血清、高纯度的Vero细胞狂犬病疫苗(PVRV-NG)正在开发中。我们先前证明,PVRV-NG暴露前预防(PrEP)具有令人满意的安全性,并且在免疫原性方面不劣于成人许可的纯化Vero细胞狂犬病疫苗。在此,我们评估了在健康成人中使用PVRV-NG与许可的人类二倍体细胞疫苗(HDCV)相比的安全性和免疫原性非劣效性(NCT01784874)。受试者接受了三次疫苗接种(第0、7和28天),12个月后,有或没有加强注射。狂犬病毒中和抗体(RVNA)在第0天、第28天(仅亚组)和第42天以及第6个月、第12个月和第12 + 14天(仅加强组)进行评估。非劣效性(第一主要目标)基于第42天RVNA滴度≥ 0.5 IU/mL(世界卫生组织血清转化标准)的受试者比例,预计阳转率≥ 99%(第二主要目标)。在每次给药后评估安全性,并在整个研究过程中进行监测。在第42天,PVRV-NG不劣于HDCV,并且达到了第一个主要目标;观察到98.3%的PVRV NG受试者和99.1%的HDCV受试者出现血清转换。由于PVRV-NG组中< 99%的受试者的RVNA滴度≥ 0.5 IU/mL,因此第二个主要目标没有达到。加强免疫对所有用PVRV-NG或HDCV组产生了RVNA滴度的强烈增加。HDCV的RVNA几何平均滴度倾向于高于PVRV NG一级疫苗接受者。在使用血清转化替代标准(在1:5血清稀释度下完全病毒中和)的补充评估中,PVRV-NG和HDCV组中分别有99.6%和100%的受试者在所有疫苗组中实现了血清转化。研究期间未观察到重大安全问题,PVRV-NG耐受性良好,在初次和加强疫苗接种后不良事件的发生率、持续时间和严重程度方面,其安全性与HDCV相似。
ClinicalTrials.gov编号:NCT01784874。
关键词:加强疫苗接种 人二倍体细胞疫苗 暴露前预防 狂犬病 Vero细胞狂犬病疫苗 无血清
1. 介绍
狂犬病是一种病毒动物传染病疾病,每年引起约59,000人死亡。它存在于150多个国家,95%的病例发生在非洲和亚洲。大多数人类感染病例发生在接触受感染动物的唾液后,通过咬、舔和抓,犬是最常见的传染源。一旦出现临床症状,没有已知的治疗方法,几乎总是导致死亡。疫苗接种是疾病预防的核心,可以作为暴露前预防(PrEP)或暴露后预防(PEP)进行。建议对暴露于狂犬病高风险的个人进行预防接种,包括:生活在狂犬病偏远地区的个人地方性动物病每年犬咬伤发生率> 5%或已知存在吸血蝙蝠狂犬病的地区;面临职业风险的个人;和可能处于危险中的旅行者。当出现以下情况时,当血清抗体浓度降至低于世卫组织规定的最低水平0.5 IU/mL,根据国家指南,在持续或频繁暴露于狂犬病高风险的人群中,建议使用暴露前加强接种。
一些基于细胞的狂犬病疫苗已经被开发并上市用于PrEP和PEP接种。人二倍体细胞疫苗(HDCV)并纯化Vero细胞狂犬病疫苗(PVRV)具有良好的安全性和有效性。基于Pitman-Moore毒株开发了新一代无血清、无抗生素的纯化Vero狂犬病疫苗(PVRV-NG)。与HDCV和PVRV不同,PVRV-NG的生产不涉及使用源自动物或人类来源的培养基成分,消除了潜在的非常规传播因子残留污染的风险,例如与牛海绵状脑病和痒病。PVRV-NG是高纯度的,低残留DNA含量(< 100 pg/剂),并且符合欧洲药典发布的标准,世界卫生组织,以及美国美国食品药品监督管理局的要求。PVRV-NG已被证明具有令人满意的安全性,并已被证明在法国健康成人中作为三剂接种方案给药时,可诱导强烈的免疫反应,并显示出与许可的PVRV相比的非劣效性,并作为中国健康成人和儿童的五剂量PEP接种。
本研究旨在对美国健康成人进行额外评估,与HDCV相比,PVRV-NG的三剂量接种方案的安全性和免疫学非劣效性,HDCV已在美国获得许可,并已在美国长期使用。
2. 方法
2.1. 研究设计和受试者
这是一项从2013年2月至2015年3月在美国六个临床研究中心进行的II期、观察者盲、对照、随机研究(clinicatrials.gov,NCT01784874)。本研究遵循《赫尔辛基宣言》和《人用药物注册技术要求协调国际会议(ICH)良好临床实践指南(GCP)》制定的标准,以及所有地方和/或国家法规和指令。知情同意是在进行任何研究程序之前从受试者处获得的。
18岁至< 65岁的健康成年人有资格入选。排除标准包括:妊娠或妊娠风险;暴露于狂犬病的高风险;既往接种过狂犬病疫苗;在首次研究疫苗接种前四周或任何研究疫苗接种后四周内接种任何疫苗;收到免疫球蛋白类或血液制品;已知或疑似先天性或获得性免疫缺陷或收到免疫抑制治疗;自报艾滋病毒、乙型肝炎或丙型肝炎血清阳性;和已知的超敏反应任何疫苗成分。
2.2. 疫苗
PVRV-NG(赛诺菲)是冻干的。每0.5 ml复溶剂量的PVRV-NG含有Wistar狂犬病病毒pitman Moore/WI 38 1503–3m菌株,效价≥ 2.5 IU (NIH效价测试)。对照疫苗,HDCV(Imovax Rabies,赛诺菲)也是冻干的,含有Wistar Rabies Virus Pitman Moore/WI 38 1503–3 M株,每复溶剂量(1 mL)的效价≥ 2.5 IU (NIH效价测试)。根据美国疾病预防中心控制、免疫实践咨询委员会(ACIP)和世界卫生组织(WHO)对暴露前接种方案的建议,在第0、第7和第28天通过肌内注射将疫苗施用到三角肌。疫苗接种在交替侧进行,例如,第一次注射在左三角肌,第二次注射在右三角肌,第三次注射在左三角肌。
2.3. 随机化和盲法
使用交互式语音应答/网络应答系统进行随机化,通过研究中心进行区块置换和分层,对于治疗分配后的第二次和第三次随机化,通过D0时分配的治疗。受试者首先以2:1的比例随机接受PVRV-NG或HDCV的一系列初级疫苗接种。进行第二次随机化,分配受试者子集(n = 120)以提供血液样品抗体反应第二次初次接种后21天的评估(第28天免疫原性子集)。第一次疫苗接种后12个月(M12),受试者第三次随机接受加强疫苗接种或不接受加强疫苗接种。在那些随机接受加强疫苗接种的人中,那些接受PVRV NG作为初次疫苗接种的人被分配到PVRV NG作为加强疫苗接种(PVRV NG/PVRV NG);而那些接受HDCV作为第一疫苗的受试者以1:1的比例被分配到PVRV-NG (HDCV/PVRV-NG)或HDCV (HDCV/HDCV)作为加强疫苗。
如同法国维尔博疫苗和HDCV在视觉上是可区分的,所以该研究是观察者盲的,即施用疫苗的人不同于评估安全性的盲的人,以避免安全性收集中的偏差。受试者不知道注射的是哪种疫苗。
2.4. 免疫原性评估
使用快速荧光灶抑制试验(RFFIT在堪萨斯州立兽医诊断实验室[KSVDL]进行,由主办方的全球临床免疫学实验室负责)。简而言之,连续稀释的血清样品与固定量的狂犬病毒的攻击标准毒株(CVS-11)和BHK-21细胞混合。然后用异硫氰酸荧光素(FITC)标记的抗狂犬病病毒核蛋白抗体,使用标准荧光显微镜记录病毒感染细胞的微观视野。使用Reed和Muench方法对50%有效剂量下的RVNA滴度进行数学转换。在用美国标准狂犬病免疫球蛋白的终点中和滴定度校准后,以每毫升国际单位(IU/mL)报告测试血清的终点中和滴定度。
主要免疫原性终点是在第42天达到RVNA滴度≥ 0.5 IU/mL的受试者百分比。次要免疫原性终点是受试者子集中所有受试者在D0和D42以及第二次给药后21天(D28)的RVNA几何平均滴度(GMT)。通过测量M6和M12的所有受试者的滴度,以及随机分组的不接受加强免疫的受试者的滴度,评估初次接种后RVNA的持久性M18还有M24。在接受加强疫苗接种的受试者子集中,在接种后2周测量RVNA滴度加强接种量(M12 + D14)以及在加强接种(M18和M24)后的第6个月和第12个月。
2.5. 安全
在每次接种疫苗后,受试者都提供了一系列日记卡,以及一把尺子和一个温度计来记录整个研究过程中的AE。
安全性结果包括:在每次注射后30分钟内报告的即时自发全身性AE;每次注射后7天内出现的请求性AE(注射部位反应、发热、头痛、不适或肌痛);从第一次注射到第三次注射后28天(D56)以及加强注射后28天的主动提供的AE。所有受试者的SAE数据收集到第12个月,接受加强接种量的受试者的SAE数据收集到第18个月。此后,收集到第24个月的死亡和相关SAE数据。
3. 统计方法
408名受试者的样本量在PVRV-NG和HDCV组分别(272名和136名)预计PVRV-NG组和HDCV组分别有230名和115名可评估受试者(假设15%不可评估)。该样本量计划在α水平为2.5%的单侧非劣效性检验中提供95%的功效,并且当基础血清转化比率(与的比例病毒中和抗体第42天的滴度≥ 0.5 IU/mL)预计为每组的99%。使用Farrington和Manning方法计算样本量。
第一个联合主要目标是证明PVRV NG在第42天(初次接种系列最后一次接种后14天)RVNA滴度≥ 0.5 IU/mL的受试者达到比例方面不劣于HDCV。如果两组间差异的双侧95%可信区间的下限高于–5 %,则认为证明了非劣效性。比例差异的95%置信区间是在没有连续性校正的情况下使用威尔逊评分法计算的。第二个共同主要目标是观察在第42天达到RVNA滴度≥ 0.5 IU/mL的受试者比例至少为99%,95%可信区间(CI)下限至少为97%。使用精确二项式(Clopper-Pearson)方法计算第二个共同主要目标的95% CI。
其他的免疫原性安全性评估是描述性的。临时后分析包括使用ACIP反应标准(即在1:5稀释度下完全血清中和的受试者比例)对主要目标进行补充评估。
主要免疫原性终点在每个方案分析集(PPAS)中进行评估,并在全分析集(FAS)中重复;报告了FAS的所有次要免疫原性终点。FAS包括接受了至少一剂初次疫苗接种系列的随机受试者,并通过接种疫苗进行分析。PPAS包括另外满足所有方案规定的纳入标准的FAS受试者,血清反应阴性(滴度低于定量下限[LLOQ;0.2 IU/mL]),正确接受了所有指定的注射,并提供了所有要求的注射血清学D0和D42的测试结果有效的样本;对PPA的分析是按分配的疫苗进行的。受试者定义了两个FAS子集,然后接受了加强接种量在第12个月(FAS-加强免疫)或那些没有接受加强免疫并在初次接种后评估抗体持久性的人(FAS-持久性)。安全性分析集(SAS)包括至少接受一剂疫苗的受试者。
4. 结果
4.1. 受试者
受试者的人口基线特征如所示表1。总体而言,410名受试者被随机分配接受初级疫苗接种(PVRV-NG,n = 273HDCVn = 137),其中408人接受了至少一剂(图1a).在M12随机抽取的183名受试者中,181名接受了a加强接种量(PVRV-NG/PVRV-NG,n = 108HDCV/PVRV-NG,n = 36HDCV/HDCV,n = 37)(图1b).总共有149名受试者被随机分组,不接受加强免疫,并随访抗体持久性(PVRV-NG,n = 112HDCV,n = 37)。PPAS包括349名受试者(PVRV-NG,n = 235[86.1%];HDCV,n = 114 [83.2%])。
表1. 初级疫苗接种治疗的基线人口统计(全分析集)。
PVRV天然气公司 | HDCV | |
第0天的平均年龄,年(SD) | 36.7 (12.0) | 37.8 (11.9) |
女性,人数(%) | 145 (53.3) | 65 (47.8) |
西班牙裔/拉丁裔,n (%) | 56 (20.6) | 30 (22.1) |
种族血统,n (%) | ||
白色的 | 186 (68.4) | 98 (72.1) |
亚洲的 | 3 (1.1) | 1 (0.7) |
黑人/非裔美国人 | 73 (26.8) | 32 (23.5) |
美洲印第安人/阿拉斯加土著 | 5 (1.8) | 0 |
夏威夷土著/其他太平洋岛民 | 3 (1.1) | 1 (0.7) |
混合起源 | 2 (0.7) | 4 (2.9) |
SD,标准差。
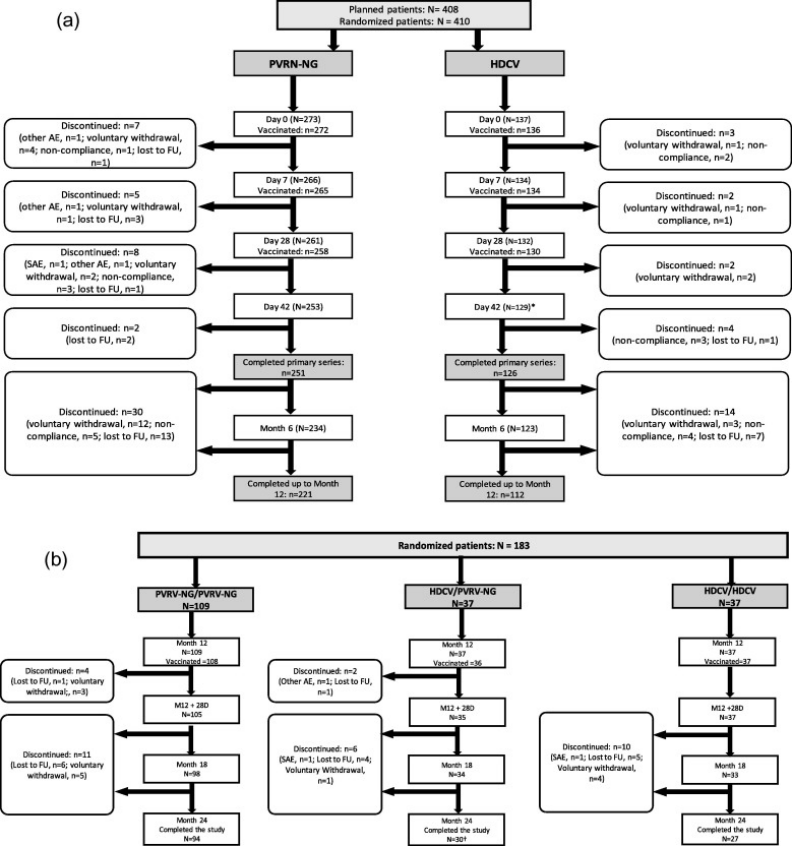
图1. a)随机初次疫苗接种系列的受试者和b)随机加强疫苗接种的受试者。*一名受试者无意中接种了PVRV-NG,而不是第三剂一级疫苗HDCV未参加D42访视,但未从研究的剩余部分中退出。一名受试者停止了加强接种,但继续进行研究,并被视为已完成研究。
4.2. 免疫原性
4.2.1. 初级疫苗接种系列
在第42天,在PPAS中,PVRV-NG组的231/235 (98.3% [95%可信区间95.7–99.5])和HDCV组的113/114 (99.1% [95%可信区间95.2–100])的受试者病毒中和抗体滴度≥ 0.5国际单位/ml。两个疫苗组之间D42 RVNA滴度≥ 0.5 IU/mL的受试者比例差异为–0.8%(95% CI–3.5,3.2),从而满足非劣效性标准。这在FAS中得到证实(比例差异为–0.8%[95% CI 3.3,2.9])。然而,五名受试者的滴度< 0.5 IU/mL:在PVRV-NG组中,两名受试者的RVNA滴度为0.4 IU/mL,两名受试者的滴度< 0.2 IU/mL,在HDCV组中,一名受试者的RVNA滴度为0.4 IU/mL。因此,第二个主要共同目标没有实现。
鉴于五名受试者的这些意外,滴度< 0.5 IU/mL,我们进行了进一步调查以确定潜在原因。我们没有发现疫苗批次、生产工艺、产品性能或特征、临床场所的血清或疫苗管理或受试者特征的任何潜在影响。在D42结果的补充分析中,使用ACIP标准血清转化(在1∶5血清稀释度下完全中和病毒),这五个受试者中的四个在1∶5稀释度下证明完全中和。总体而言,在第42天,PVRV-NG组和HDCV组分别有99.6%和100%的受试者采用ACIP标准进行血清转换,两组之间的比例差异为–0.4%(95% CI–2.22,2.56)。由于PVRV-NG组完全血清中和的受试者比例的95%可信区间下限大于97.5%,HDCV组大于100%,因此这两个主要结果指标都已通过这些指标得以实现临时后标准(补充表S1)。
在FAS中,PVRV-NG和HDCV组的接种前(D0)GMT分别为0.101 IU/mL和0.104 IU/mL;只有三个受试者(PVRV-NG,n = 1;HDCV,n = 2)的滴度高于测定的LLOQ(> 0.2 IU/mL)。三剂(D42)后,PVRV-NG组和HDCV组的RVNA GMT分别增加到10.5 IU/mL和16.8 IU/mL。在FAS的D28受试者亚组中,PVRV-NG组和HDCV组分别有71/80 (88.8%)和43/43 (100%)的受试者在两次给药后RVNA滴度≥0.5 IU/mL;D28的GMT为2.35 IU/mL (PVRV-NG)和5.94 IU/mL (HDCV)。
4.2.2. 抗体持久性和加强疫苗接种
两组初次接种疫苗后,滴度≥ 0.5 IU/mL和平均RVNA滴度的受试者比例在M6和M12时均下降,PVRV NG组在这两个时间点的水平均低于HDCV组(图2a和2b)。在第12天,PVRV-NG组和HDCV组分别有28.3%和54.1%的受试者的滴度≥0.5 IU/mL;两组在M12时的GMT分别为0.214和0.409。

图2. 免疫反应和抗体持久性在受试者中的比例病毒中和抗体滴度≥ 0.5 IU/mL和受试者的GMT概况(全分析集-持久性和加强疫苗接种子集)。数据为95%置信区间,时间点和“n”显示在图下方。
在PVRV-NG组和HDCV组中,随机选择不接受加强接种量的受试者中,在M18时,达到滴度≥ 0.5 IU/mL的百分比分别为31.1%和52.9%;在M24时,分别为34.4%和46.9%,平均RVNA滴度在两个时间点都保持较低(图2a和2b)。对于在第12天接受加强接种量的受试者,两周后所有加强接种量组的RVNA滴度均显著增加(GMT:PVRV-NG/PVRV-NG,31.2;HDCV/PVRV-NG,39.2;HDCV/HDCV,59.8);所有受试者在加强免疫后两周内RVNA滴度均≥ 0.5 IU/mL(图2a和2b)。在初次疫苗接种系列后第42天达到RVNA滴度< 0.5 IU/mL的五名受试者中的三名被包括在随机接受加强免疫的受试者中。在加强接种量后两周,所有这三名受试者的RVNA滴度都远远超过0.5 IU/mL;PVRV-NG组中的两名,在第42天滴度< 0.2 IU/mL和0.4 IU/mL,加强后滴度分别为3.7和4.3 IU/mL;HDCV组中的一名受试者在第42天的滴度为0.4 IU/mL,加强免疫后的滴度为4.5 IU/mL。
接受HDCV加强免疫的受试者组的GMT往往高于接受PVRV-NG加强免疫的受试者组:加强免疫后14天,接受HDCV加强免疫的受试者的GMT为59.8 (95% CI 38.7,92.6),而在PVRV-NG或HDCV初次接种后,接受PVRV-NG加强免疫的受试者的GMT分别为31.2 (95% CI 25.2,38.6)和39.2 (95% CI 27.5,55.8)。
4.2.3. 安全
初次和加强疫苗接种后的安全性结果总结于表2。总的来说,在研究过程中没有观察到重大的安全性问题。在任何初次疫苗剂量后,两组均没有立即出现疫苗相关AE。请求的注射部位反应PVRV-NG组(36.1%)比HDCV组(62.4%)首次接种疫苗的频率低;最常报告的是注射部位疼痛大多数为1级严重程度,并在接种疫苗后3天内解决。两组中诱发的全身反应的报告率相同。PVRV-NG组中的13名(4.9%)受试者和HDCV组中的4名(3.0%)受试者报告了至少一次3级全身反应;所有问题都自行解决或通过药物解决。两组间报告的主动非严重AE和ar频率相同(分别为31.6%和4.4%),大多数为1级或2级严重程度。两组均无被认为与疫苗接种相关的SAE报告。HDCV组中有一例非严重AE被评估为与研究疫苗相关,并导致研究中止(第一次接种疫苗后出现1级瘙痒和全身皮疹)。
表2. 在任何初次疫苗剂量和加强疫苗接种后7天内引发的不良事件(安全性分析集)。显示的数据是经历≥ 1次事件的受试者人数(%)。
初次疫苗接种(任何剂量) | |||
PVRV-NG (N = 266) | HDCV (N = 133) | ||
至少1个请求反应 | 174 (65.4) | 98 (73.7) | |
注射部位反应 | 96 (36.1) | 83 (62.4) | |
疼痛 | 94 (35.3) | 81 (60.9) | |
红斑 | 1 (0.4) | 6 (4.5) | |
膨胀 | 2 (0.8) | 5 (3.8) | |
全身反应 | 154 (57.9) | 77 (57.9) | |
发热 | 3 (1.1) | 3 (2.3) | |
头痛 | 113 (42.5) | 51 (38.3) | |
不舒服 | 80 (30.1) | 49 (36.8) | |
肌痛 | 105 (39.5) | 60 (45.1) | |
加强疫苗接种 | |||
PVRV天然气公司 / PVRV天然气公司 | HDCV / PVRV天然气公司 | HDCV / HDCV | |
至少1个请求反应 | 47 (45.2) | 16 (44.4) | 14 (37.8) |
注射部位反应 | 21 (20.2) | 8 (22.2) | 11 (29.7) |
疼痛 | 21 (20.2) | 8 (22.2) | 11 (29.7) |
红斑 | 1 (1.0) | 0 (0) | 1 (2.7) |
膨胀 | 0 (0) | 0 (0) | 0 (0) |
全身反应 | 36 (34.6) | 14 (38.9) | 13 (35.1) |
发热 | 5 (4.9) | 1 (2.8) | 0 (0) |
头痛 | 29 (27.9) | 12 (33.3) | 7 (18.9) |
不舒服 | 19 (18.3) | 7 (19.4) | 6 (16.2) |
肌痛 | 24 (23.1) | 7 (19.4) | 12 (32.4) |
n,治疗组中包括的受试者数量。
加强免疫接种后,PVRV-NG加强组(总体20.7%)报告的请求注射部位反应比HDCV加强免疫组(29.7%)少;对于三个加强组中的每一个,几乎所有的都是注射部位疼痛,大多数是1级严重程度,并且大多数在接种后3天内解决。据报道,在所有加强治疗组中,诱导全身反应的频率相似。五名受试者报告了12例3级系统性反应(1例发热、4例头痛、4例不适和3例肌痛),所有受试者均接受了PVRV-NG加强接种。11.1% (PVRV-NG)和10.8% (HDCV)的受试者报告了主动提供的非严重AE;大多数为1级或2级全身事件,与疫苗接种无关。PVRV-NG组中有一例不请自来的非严重不良事件(注射部位出血)被认为与疫苗接种有关。在加强免疫后,没有出现被认为与疫苗接种相关的SAE。
5. 讨论
本研究检测了成人在暴露前免疫中注射PVRV-NG与HDCV相比的安全性和非劣效性。我们之前的研究表明,PVRV-NG的PrEP在免疫原性上不逊于法国维尔博疫苗在成人中以及儿童的HDCV。目前的研究表明PVRV-NG与HDCV相比在以下方面具有非劣效性血清转化最后一次初级预防接种后14天的比率。然而,PVRV-NG组中的四名受试者的滴度< 0.5 IU/mL(包括两名滴度为0.4 IU/mL的受试者),因此发展为病毒中和抗体D42时滴度≥0.5 IU/mL(98.3%)低于满足第二个共同主要目标所需的99%临界值。HDCV组的相应比例为99.1%(一名受试者在第42天的滴度< 0.5 IU/mL [0.4 IU/mL])。
考虑到HDCV在超过40年的临床使用中所表现出的完善的保护性免疫反应,尽管表现出相对于HDCV的非劣效性,但PVRV-NG疫苗接种后的血清转换率不符合预先定义的标准这一发现是出乎意料的。事实上,这两种疫苗的效价≥ 2.5 IU (NIH效价测试),这是在人体内提供足够免疫原性的最低要求。此外,这些结果与之前描述的用PVRV-NG观察到的保护性免疫反应不一致或HDCV,或与儿童的近期结果。我们对疫苗批次、临床点的血清样本管理或受试者特征的额外调查没有解释观察到的结果。然而,一些受试者对狂犬病疫苗接种的抗体反应差的现象并不是前所未有的,先前描述的不同市售疫苗的PrEP或PEP后的病例。还应该考虑到,PrEP的主要目标是确保足够的启动,以便能够快速有效地回忆免疫记忆在暴露于病毒的情况下,从而避免需要狂犬病免疫球蛋白,价格昂贵且难以获得。在目前的研究中,在第42天RVNA滴度< 0.5 IU/mL的五名受试者中有三名是随机接受PVRV-NG或HDCV的加强接种量;在加强免疫后两周,所有三种疫苗都表现出足够的反应,滴度超过0.5 IU/mL,表明这些受试者的有效免疫引发。评估RVNA的测试的可变性可能在这些受试者中观察到的滴度< 0.5 IU/mL中发挥了作用。为了进一步研究RFFIT检测性能的潜在可变性,进行了独立的实验室分析(巴斯德研究所,巴黎和亚特兰大卫生协会),以重新检测初始检测中D42 RVNA滴度低于世卫组织血清转化阈值(< 0.5 IU/mL)的样本子集,以及选择RVNA滴度≥ 0.5 IU/mL的疫苗接种后样本和RVNA滴度低于LLOQ的疫苗接种前样本。那些在初始测试中滴度> 0.5 IU/mL的样品保持> 0.5 IU/mL,接种前样品保持低于LLOQ在第42天初次检测时滴度< 0.5 IU/mL的受试者中,除一人(PVRV-NG组)外,所有受试者再次检测时RVNA滴度均≥ 0.5 IU/mL。虽然我们没有发现任何个体因素导致了观察到的变异,但变异的影响似乎对那些滴度在阈值附近的人最大。我们还使用替代的ACIP标准对所有样品进行了补充分析(在1:5稀释度下完全中和[28]).。当应用ACIP标准时,99.6%的PVRV NG受试者在第42天达到血清转换,包括使用预先定义的研究标准未能达到血清转换的4名受试者。这提供了证据,表明这四名受试者的疫苗接种产生了足够的引发,并在随后接受加强免疫的三名受试者中得到证实。
无论受试者接种后RVNA滴度≥ 0.5 IU/mL,对PVRV-NG或HDVC初次疫苗接种系列反应的加强稳定性对于确保实现PrEP的目标可能很重要,特别是在2018年世卫组织推荐的新的较短PrEP方案之后以及美国的ACIP。观察到的抗体反应的强度在PVRV-NG后倾向于低于HDCV后。虽然其原因尚不清楚,但一种假设可能是,当用酶联免疫吸附测定在HDCV批次中高于PVRV-NG批次。这种观察到的滴度大小的差异不太可能具有临床意义,因为在所有组中加强免疫后,RVNA滴度都远远超过0.5 IU/mL。事实上,我们的发现,加上以前的观察,即100%的受试者在初始系列或单次PVRV-NG加强接种量后血清转化,表明当作为暴露前方案施用时,PVRV-NG将诱导足够的免疫反应以提供保护。这项研究是在世卫组织2010指南推荐3剂量暴露前免疫方案时进行的。最近提出的2剂量方案及其相关的加强接种量未在此评估。因此,这项研究提供了证据,证明PVRV-NG不低于使用3剂肌肉注射制剂的护理标准,并证实了可加强性。
总之,这项研究表明,就血清转换率而言,PVRV-NG的PrEP方案并不劣于已获许可的HDCV。PVRV-NG组的4名受试者和HDCV组的1名受试者在预接种疫苗后未达到RVNA滴度≥ 0.5 IU/mL,导致PVRV-NG组RVNA滴度≥ 0.5 IU/mL的受试者比例(< 99%)不足以达到第二个主要免疫原性目标。然而,我们显示在所有组中加强疫苗接种后,RVNA滴度显著增加,证实了初始疫苗接种系列的引发效应和PVRV NG的加强稳定性。重要的是,PVRV-NG得到了很好的容忍,有越来越少被邀请的趋势注射部位反应比HDCV组的要高。没有观察到意外的安全性信号,这与早期对PVRV-NG的安全性分析一致。
6. 数据共享声明
合格的研究人员可以要求访问患者水平数据和相关研究文件,包括临床研究报告、研究方案(包括任何修订版)、空白病例报告表、统计分析计划和数据集规格。患者水平的数据将被匿名,研究文件将被编辑以保护试验受试者的隐私。有关赛诺菲数据共享标准、合格研究和请求访问流程的更多详情,请访问:https://www.vivli.org/.
来源:Vaccine
. 2022 Jun 28;S0264-410X(22)00806-4. doi: 10.1016/j.vaccine.2022.06.040. Online ahead of print.
Safety and immunogenicity of a serum-free purified Vero rabies vaccine in healthy adults: A randomised phase II pre-exposure prophylaxis study
https://m.sciencenet.cn/blog-55647-1346002.html
上一篇:213 人源狂犬病单抗体混合制剂可广泛中和北美狂犬病病毒变异株 ——人源单抗是狂犬病暴露后预防的有希望的候选药物
下一篇:215 狂犬病暴露后单克隆抗体鸡尾酒疗法