博文
将GO、Pathway富集结果整合在一张高颜值圆圈图上
|
富集分析是生物医学论文中非常常见的一类分析,例如GO富集分析,Pathway富集分析等。其结果一般包括以下几个要素:1,名字(GO term或者KEGG description);2,该名字所包含的基因数目;3,该名字所包含的基因与进行富集分析的输入基因的重叠数目;4,富集的P值、FDR值等。富集结果的常见绘图方式包括:气泡图,条形图,弦图等。今天我们利用circlize R包整合富集结果,绘制一个高大上的圈图。

图1.GO、Pathway富集圈图
如图1所示,从外到内共4个track,中间的为图例(从上到下依次为:分类,p值,上调或者下调)
1)分类track
相同颜色的为同一分类,例如黄色表示BP,蓝色表示MF,最外圈刻度表示总的基因数,10的次方表示。总的基因数即图2中所有黄色圈所包含的基因去重后的基因数,或者推荐使用比所有条目中基因数最大值更大的一个值(一般设置为整百或者整千)。
2) term里边的基因数
矩形长度表示该term中包含的基因数(例如GO:0031625这个term中包含306个基因),与最外圈的刻度成比例。矩形的颜色表示该term的富集p值(或者FDR值,q值等,经过-log10转化),颜色越深表示P越小。
3)重叠基因数
矩形长度表示该term中包含的基因与进行基因富集分析时所输入的基因的重叠基因数。例如进行富集分析的基因有500个,其中与GO:0031625中306个基因重叠的基因为38个。由于这个数字一般较小,因此矩形的长度与该track中最的基因数成比例。一般我们要么使用up的基因,要么使用down的基因进行富集分析,因此这里仅一种颜色。
4)富集分数bar
Bar的高度即第三个track里的基因数占第二个track里对应基因数的比例(如图2所示)。数值范围为0到1,这里每个小圈表示0.2。
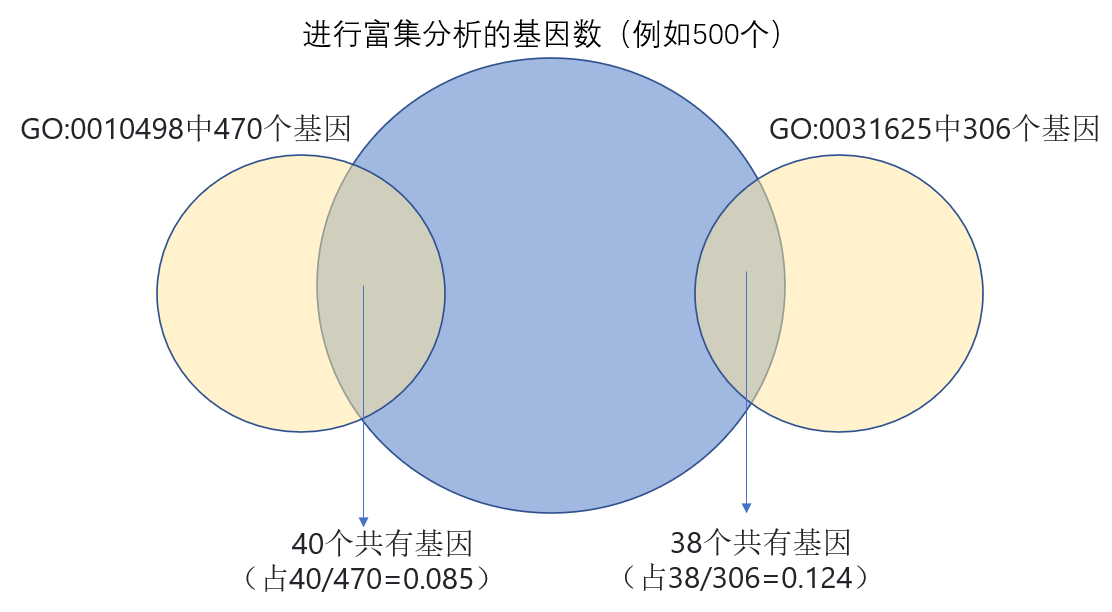
图2. 基因数及比例来源示意图
1, 打开绘图页面
首先,使用浏览器(推荐chrome或者edge)打开富集圈图绘制页面。左侧为常见作图与分析导航,中间为数据输入框和可选参数,右侧为描述和结果示例。也可以在主页搜索框中搜索GO,找到绘图页面。
http://www.bioinformatics.com.cn/plot_basic_go_pathway_circlize_plot_140
图3.富集圈图绘图页面
2,示例数据
点击右侧“示例数据”链接下载excel格式的示例数据。
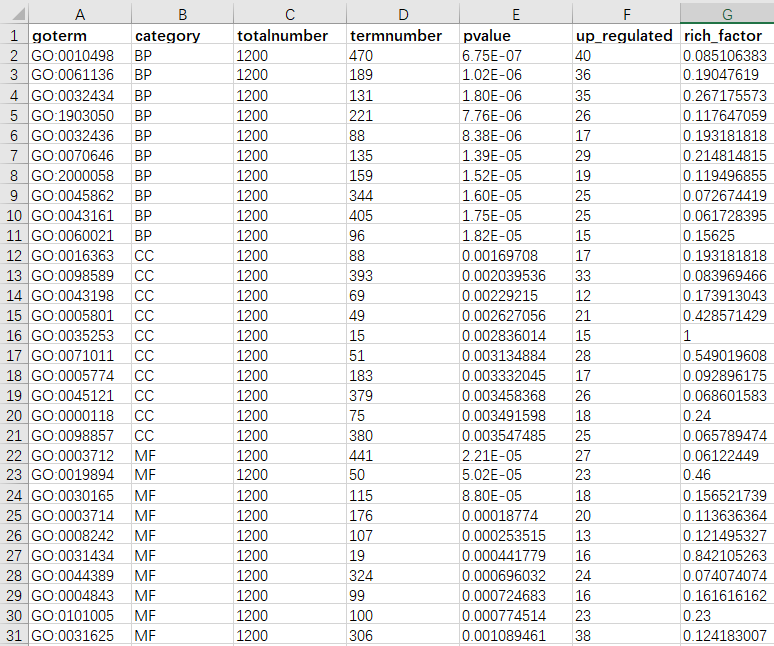
图4. 输入数据示例
示例数据(仅供参考)包括7列:
第1列:GO或者KEGG条目ID。推荐使用ID,因为名字长短不一,很难统一。
第2列:分类名。可以包含1类,2类,3类,或者4类,例如单独绘制KEGG通路富集结果;同时绘制GO-BP,KEGG;同时绘制BP、CC、MF;甚至同时绘制BP、CC、MF、KEGG。同一分类放在一起,不要相互穿插。
第3列:总基因数。总基因数即图2中所有黄色圈所包含的基因去重后的基因数,或者推荐使用比所有条目中基因数最大值更大的一个值。例如第4列中最大的数值是470,这里可以使用1200,用来从整体上控制第二个track的矩形长度,是一个比较松散的值(一般设置为整百或者整千)。
第4列:数据库中某term包含的基因数。在使用同一个数据库版本的情况下,这个数值是固定不变的,例如GO:0010498里边就包含470个基因。
第5列:某term富集的p值(或者q值,FDR值等)。
第6列:做富集分析时输入的基因与该term中包含基因的重叠基因数(见图2)。
第7列:第6列与第4列的比值(见图2)。
3,粘贴示例数据
直接复制示例数据中的A-G列数据,然后粘贴到输入框。
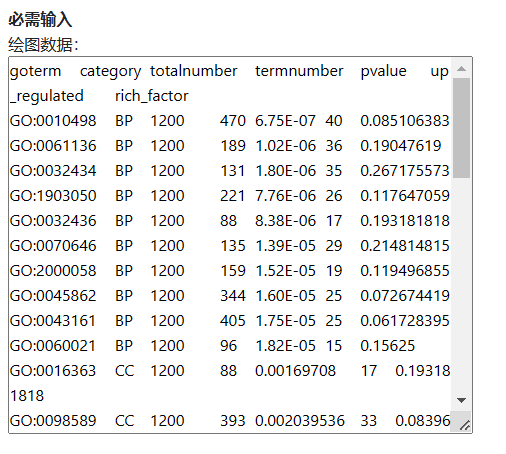
图5. 必需输入
注意:不是拷贝excel文件,是拷贝excel文件里边的数据。另外粘贴到输入框后,格式乱了没关系,只要在excel中是整齐的就行。并且数据矩阵中不能有空的单元格,中文字符等。
4,修改参数,并提交
我们设置了图片尺寸,字体大小,颜色,字体类型等参数,基本能满足日常绘图使用。如需更高级的定制,请联系我们。
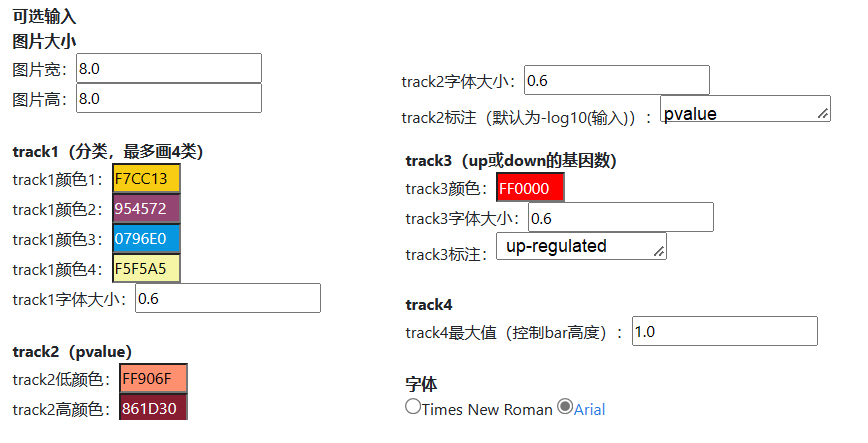
图6.字体、颜色等可调参数
5,提交出图
粘贴好输入数据,调整好参数(或者全部默认)后,点击提交按钮,约8秒后,会在页面右侧出现预览图。我们提供了4种图片格式供下载使用,两种矢量图(pdf,svg)和两种标量图(600 dpi tiff和300 dpi png)。
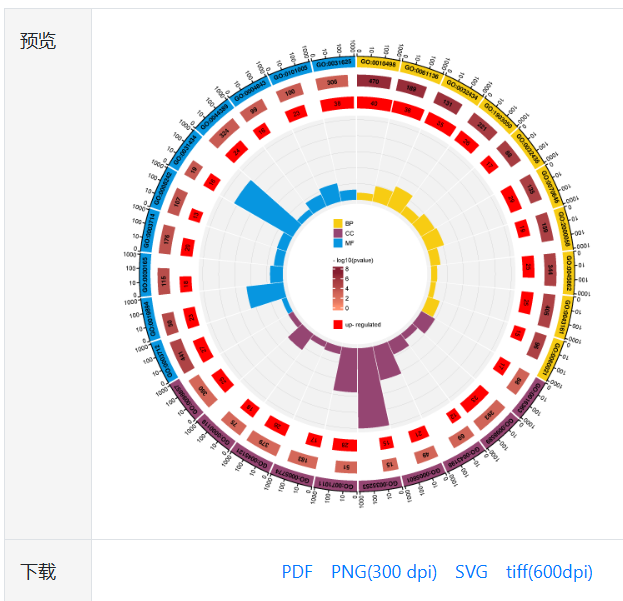
图7.预览与下载
Tips:
1,该图最大限度地利用有限的空间展示更多的数据内容。因此条目不能过多,推荐总共30个以内的GO和KEGG条目,否则太拥挤。
2,第4个track最大值默认为1.0,可以修改成其他值,以整体上降低或者降低柱子的高度。
3,一般我们使用up或者down的基因做富集分析,对于使用up+down富集结果不适合这个图。
4,对于DAVID等富集分析结果,各列的名字可能不一样,需要根据数值代表的意思调整成示例数据即可。
没有预览就是没有出图,这时请参考示例数据,检查自己输入数据的格式。
遇到文字截断,需要修改字体、调整字体大小等,使用科研作图,用inkscape编辑svg矢量图
微生信助力高分文章,用户66000+,引用870+
https://m.sciencenet.cn/blog-707141-1371697.html
上一篇:在线计算lncRNA-mRNA共表达相关系数,并使用cytoscape绘制共表达网络图
下一篇:如何查找“铁代谢”相关的基因?