ВЉЮФ
QIIME 2НЬГЬжЎЩњГЩЬиеїБэКЭЬиеїађСа(АИР§Жў)
|
АИР§ЖўЃЈЦфЫћfastqЪ§ОнИёЪНЃЈЕЅЖЫЪ§ОнЃЉЃЉ

ЃЈвЛЃЉбљБОдЊЪ§Он
дкПЊЪМШЮКЮЗжЮіжЎЧАЃЌЪьЯЄдЊЪ§ОнКмживЊЁЃдЊЪ§Онmetadata.tsvЁЃ
qiime metadata tabulate --m-input-file metadata.tsv --o-visualization metadata.qzv |

ЁОБИзЂЁПЃЃq2_typeжИСюПЩвджИЖЈЗжРрЪ§ОнРраЭ

ЃЈЖўЃЉбљБОдЊЪ§Он
НЬГЬНЋЪЙгУбљБОЧхЕЅИёЪН(manifest format)ЕМШыађСаЃЌетЪЧвЛжждкQIIME 2жаЕМШыВ№ЗжбљБОЪ§ОнЕФЭЈгУЗНЗЈЁЃЦеЭЈгУЛЇГЃгУЕФЯТЛњЪ§ОнИёЪНЮЊ.fastqЮФМўЃЌашвЊДДНЈвЛИіЧхЕЅЮФМўЃЌШЛКѓЪЙгУqiime tools importУќСюЪжЖЏЪфШыЁЃЧхЕЅЮФМўЪЧвЛИіЮФБОЮФМўЃЈ.tsvЛђ.txtИёЪНЃЉЃЌЫќНЋЪОР§БъЪЖЗћгГЩфЕНfastq.gzЛђfastqЕФОјЖдЮФМўТЗОЖЃЌЦфжаАќКЌЪОР§ЕФађСаКЭжЪСПЪ§ОнЁЃЧхЕЅЮФМўЛЙжИЪОУПИіfastq.gzЛђfastqЮФМўжаЕФЖСШЁЗНЯђЁЃfastq.gzЮФМўЮЛжУЕФОјЖдЮФМўТЗОЖПЩвдАќКЌЛЗОГБфСПЃЈР§Шч$PWDЃЉЁЃ
ЧхЕЅЮФМўmanifest.tsvФкШнЃЈЕЅЖЫЪ§ОнЃЉЃК
![]()
ЪЙгУЮФМўЧхЕЅЕМШыЪ§ОнЃК time qiime tools import\ --type "SampleData[SequencesWithQuality]"\ --input-format SingleEndFastqManifestPhred33V2\ --input-path manifest.tsv\ --output-path demux_seqs.qza НсЙћПЩЪгЛЏЃК time qiime demux summarize --i-data demux_seqs.qza --o-visualization demux_seqs.qzv ЪфГіНсЙћЮФМўЃКdemux_seqs.qzv ЁОБИзЂЁПЪЙгУqiime demux summarizeУќСюМьВщбљБОЕФађСаКЭВтађЩюЖШЃЈЫќЬсЙЉУПИібљБОжаађСаЪ§МАађСажЪСПЕФаХЯЂЃЉ |
НсЙћеЙЪОЃК


ЁОБИзЂЁПЕМШыЪ§ОнЯъМћЃКПЦбЇЭјЁЊQIIME 2НЬГЬжЎЪ§ОнЕМШыЃЈimporting dataЃЉ - СѕЪїЧрЕФВЉЮФ (sciencenet.cn)

ЃЈШ§ЃЉађСажЪСППижЦгыЩњГЩЬиеїБэКЭЬиеїађСа
QIIME 2ВхМўЖржжжЪСППижЦВЂЩњГЩЬиеїБэЕФЗНЪНжївЊгаСНжжЃЌвЛжжЪЧЭЈЙ§ШЅдыЃЌМДЩњГЩРЉді/ОјЖдађСаБфЬхЃЈAbsolute Sequence VariantsЃЌASVЃЉЃЌASVЪЧзюНќЗЂеЙЕФаТвЛДњЗНЗЈЃЌдкЙІФмЩЯЬсЙЉИќКУЕФЗжБцТЪЁЃASVПЩвдЛљгк400bpЛђИќЖрађСажаЕЅИіКЫмеЫсЕФВювьРДЗжРыЬиеїЃЌЩѕжСГЌЙ§99ЃЅЭЌвЛадOTUОлРрЕФЗжБцТЪЁЃФПЧАдкQIIME 2 жаПЩЭЈЙ§DADA2ЃЈq2-dada2ЃЉКЭDeblurЃЈq2-deblurЃЉВхМўЪЕЯжЁЃЕкЖўжжЪЧЭЈЙ§ОлРрЩњГЩВйзїЗжРрЕЅдЊЃЈOperational Taxonomic UnitsЃЌOTUЃЉЃЌетжжЗНЗЈзд2010ФъвдРДБуЕУЕНСЫЙуЗКгІгУЁЃQIIME 2ФПЧАПЩЭЈЙ§q2-vsearchВхМўЪЕЯжЁЃСНжжЗНЗЈВЛЭЦМізщКЯЪЙгУЁЃ
ЁОВЙГфЁПOTU(Operational Taxonomic Units)ЃКЪЧЭЈЙ§вЛЖЈОрРыМЦЫуСНСНВЛЭЌађСажЎМфЕФОрРыЖШСПКЭЯрЫЦадЃЌЩшжУЬиЖЈЕФуажЕЃЌЛёЕУЭЌвЛуажЕЯТЕФОрРыОиеѓЃЌНјааОлРрВйзїЃЌаЮГЩВЛЭЌЕФЗжРрЕЅдЊЁЃ
БОНЬГЬНЋзХжиНщЩмDADA2КЭDeblurСНжжЗНЗЈЁЃ
ЗНЗЈвЛЃКDADA2
ЖўДњВтађЕФДэЮѓЪЧЫцЛњЗЂЩњЕФЃЈМДЃЌШЮвтСНЬѕађСаЕФВтађДэЮѓЯрЖдЪЧЫцЛњЗЂЩњЕФЃЌвЛЬѕађСаЕФШЮвтСНИіЮЛжУЕФВтађДэЮѓвВЪЧЫцЛњЗЂЩњЕФЃЌВЛДцдкЙиСЊадЃЉЁЃDADA2жЪСППижЦЙ§ГЬНЋЙ§ТЫЕєдкВтађЪ§ОнжаМјЖЈЕФШЮКЮphiXађСаЃЌВЂЭЌЪБЙ§ТЫЧЖКЯађСаЃЈМДЧЖКЯЛљвђЃЌОЭЪЧСНИіЛљвђЙВгУвЛЖЮDNAађСаЃЌетСНИіЛљвђГЦЮЊЧЖКЯЛљвђЃЉЁЃдкDADA2жаЃЌЫЋЖЫКЯВЂЃЌШЅГ§ЧЖКЯЬхЃЌНиШЅНгЭЗађСаНЕдыЩњГЩfeature tableЖМЪЧвЛВНЭъГЩЕФЁЃ
ЁОВЙГфЁПphiXађСаЭЈГЃДцдкгкБъМЧЛљвђIlluminaВтађЪ§ОнжаЃЌгУгкЬсИпРЉдізгВтађжЪСПЁЃзюжївЊЕФФПЕФЃК1ЁЂЕїНкМюЛљЦНКтЃЌИФЩЦВтађвЧЕФПеМфаЃе§ЃЌБугкКѓЦкЬсИпbase callingЕФзМШЗадЃЛ2ЁЂгЩгкPhixађСавбжЊЛљвђзщНЯаЁЃЌдкВтађЕФЙ§ГЬжаIlluminaЕФВтађвЧОЭПЊЪМНЋВтЕФreadгыphixЛљвђзщНјааБШНЯЃЌдЄЙРВтађжИБъЁЃ
ЁОБИзЂЁПдЫааDADA2жЎЧАвЊШЗБЃВтађЪ§ОнТњзувдЯТЙцЗЖЃК
ЃЈ1ЃЉбљЦЗвбБЛВ№ЗжКУЃЌМДУПИібљЦЗвЛИіfq/fastqЮФМўЃЈЛђепЫЋЖЫГЩЖдfqЮФМўЃЉЃЛ
ЃЈ2ЃЉвбОШЅГ§ЗЧЩњЮяКЫЫсађСаЃЌБШШчЃКв§ЮяЃЈprimersЃЉЃЌНгЭЗЃЈadapters or barcodesЃЉЃЌlinkerЕШЃЛ
ЃЈ3ЃЉШчЙћбљЦЗЪЧЯТЛњЕФЫЋЖЫВтађЃЌЦфгІОпгаЫЋЖЫВтађЕФЯрЦЅХфЕФСНИіfqЮФМўЁЃ
ЪЙгУDADA2ВхМўНјаажЪСППижЦЃК time qiime dada2 denoise-single | --p-trim-leftЃКНиШЁзѓЖЫЕЭжЪСПађСаЁЃгУгкЧаГ§ЕЭжЪСПађСаЁЂbarocdeЛђв§ЮяЁЃ --p-trunc-lenЃКађСаНиШЁГЄЖШЃЌвВЪЧЮЊСЫЧаГ§гаЖЮЕЭжЪСПађСаЁЃвЛАуДгађСажЪСППЊЪМДѓЗљЖШЯТНЕЕФЮЛжУПЊЪМЧаГ§ЁЃ |
| ЁОУќСюзЂЪЭЁП ЃЈ1ЃЉдкЪЙгУqiime dada2 denoise-single/ qiime dada2 denoise-pairedЪБПЩЩшжУ--p-n-threads ВЮЪ§ЃЌгУгкЩшжУдЫааЪБЪЙгУЕФЯпГЬЪ§СПЁЃЯпГЬдНЖрЃЌдђдЫааЫйЖШдНПьЁЃЕБЯпГЬЩшжУЮЊ0ЪБдђФЌШЯЪЙгУШЋВПЯпГЬЃЛ ЃЈ2ЃЉ--p-trim-leftНиШЁзѓЖЫЕЭжЪСПађСаЃЌгаЪБгУгкЧаГ§ЕЭжЪСПађСаЁЂbarocdeЛђв§ЮяЁЃВщПДdemux_seq.qzvЮФМўжаЕФЯфЯпЭМЃЌзѓЖЫжЪСПЖМКмИпЃЌЮоЕЭжЪСПЧјЃЌЩшжУЮЊ0ЃЛЛђПЩжБНгКіТдДЫВЮЪ§ЩшжУЃЛ ЃЈ3ЃЉ--p-trunc-lenађСаНиШЁГЄЖШЃЌвВЪЧЮЊСЫШЅГ§гвЖЫЕЭжЪСПађСаЃЌЮвУЧПДЕНДѓгк150вдКѓЃЌжЪСПЯТНЕМЋДѓЃЌЩѕжСжаЮЛЪ§ЖМЯТНЕжС20вдЯТЃЌашвЊШЋВПШЅГ§ЃЌзлКЯПМТЧОіЖЈЩшжУЮЊ150ЃЛ ЃЈ4ЃЉЕБДІРэЫЋЖЫЪ§ОнЪБЃЌашПМТЧНиШЁКѓЕФађСаЪЧЗёПЩвдГЩЙІЦДНгЁЃФПЧАзюЖЬЕФЦДНгГЄЖШЮЊв§ЮяГЄЖШ+12bpЁЃ |
* ЭГМЦНсЙћПЩЪгЛЏ
qiime metadata tabulate --m-input-file dada2_stats.qza --o-visualization dada2_stats.qzv |
ФкШнЮЊУПИібљБОЕФЪфШыЁЂЙ§ТЫЁЂШЅдыКЭЗЧЧЖКЯЬхЕФЭГМЦНсЙћЁЃеЙЪОСЫбљБОЕФжЪСППижЦНсЙћЃЌгУгкбљБОвьГЃЩИбЁКЭЬиеїБэГщЦНБъзМЛЏЁЃ
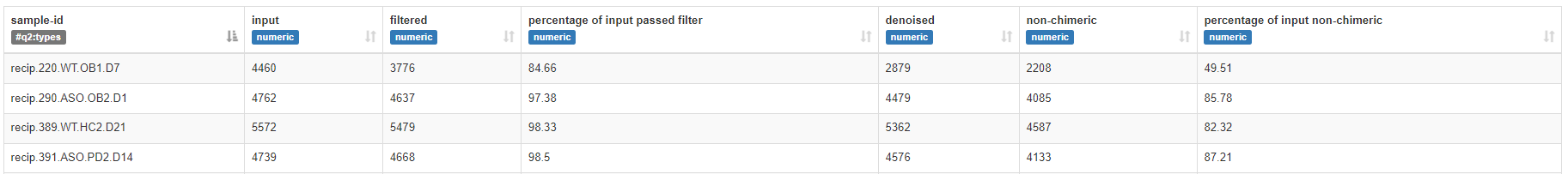
* ЬиеїБэПЩЪгЛЏ
qiime feature-table summarize --i-table dada2_table.qza --o-visualization dada2_table.qzv --m-sample-metadata-file metadata.tsv |
ЯТЭМеЙЪОСЫбљБОЪ§ЁЂЬиеїЪ§КЭЗжВМЕШаХЯЂЁЃ

ЯТЭМеЙЪОСЫУПИібљБОжаЕФЬиеїЪ§ФП

ЯТЭМЬиеїаХЯЂЃЌЗжБ№ЮЊЬиеїУћГЦЁЂГіЯжЦЕТЪКЭЙВГіЯждкЖрЩйИібљБОжаЁЃ

* ДњБэађСаПЩЪгЛЏ
qiime feature-table tabulate-seqs --i-data dada2_rep_set qza --o-visualization dada2_rep_set.qzv |
ЯТЭМеЙЪОДњБэађСаЭГМЦаХЯЂЁЃгаГЄЖШеЊвЊЁЂВЛЭЌАйЗжБШЯТЕФГЄЖШЭГМЦКЭОпЬхДњБэађСаМюЛљаХЯЂ

ЗНЗЈЖўЃКDeblur
PCRКЭВтађЙ§ГЬжаЕФдывєЯожЦСЫЧјЗжИќЯрНќЕФЮяжжЁЃвЛаЉЬиЪтЕФЩњЬЌгІгУгыПЦбЇбаОПашвЊИќОЋШЗЕФЮяжжЧјЗжЁЃвђДЫЃЌЬсГіСЫDeblurШЅдыЕФЗНЗЈЁЃDeblurЗНЗЈЬсГіСЫsub-operational-taxonomic-unit (sOTU) ЬсГіИќОЋШЗЕФЗжРрбЧOTUЕФИХФюЃЌДЫИХФюгыASVвтвхЯрЭЌЃЌжЛЪЧУћзжВЛЭЌЁЃ
ЁОБИзЂЁПdeblurФПЧАжЛФмЖдЕЅЖЫађСаНјааШЅдыЁЃШчЙћЬсЙЉФЉКЯВЂЕФЫЋЖЫађСаЮЊЪфШыЃЌНЋЖдЗДЯђађСаВЛзїШЮКЮВйзїЁЃЧызЂвтЃЌdeblurНгЪмКЯВЂЕФађСаЃЌВЂНЋЫќУЧЪгЮЊЕЅЖЫађСаЃЌвђДЫШчЙћЪЙгУdeblurНјааШЅдыЃЌашвЊЯШКЯВЂЖСШЁЁЃ
ЕквЛВН АДВтађМюЛљжЪСПЙ§ТЫађСа
time qiime quality-filter q-score --i-demux demux_seqs.qza --o-filtered-sequences demux-filtered.qza |
ЕкЖўВН deblurШЅды16SЙ§ГЬЃЌЪфШыЮФМўЮЊжЪПиКѓЕФађСаЃЌЩшжУНиШЁГЄЖШВЮЪ§ЃЌЩњГЩНсЙћЮФМўгаДњБэађСаЁЂЬиеїБэЁЂбљБОЭГМЦ
time qiime deblur denoise-16S |
ПЩЪгЛЏЪфГіЮФМўЃЌКЭdada2НсЙћРрЫЦ: МюЛљжЪСПЙ§ТЫЭГМЦНсЙћ |
DeblurОпгавдЯТЬиЕуЃК
ЃЈ1ЃЉЪЙгУЮѓВюЗжВМРДЛёЕУМйЖЈЕФЮоЮѓВюађСаЃЛ
ЃЈ2ЃЉМѕЩйСЫМЦЫуЕФашЧѓЃЌЕУЕНСЫИќИпЕФЬивьадКЭУєИаадЃЛ
ЃЈ3ЃЉжЛЪмРЉдіађСаЖСГЄКЭЖрбљадЕФЯожЦЃЛ
ЃЈ4ЃЉПЩвддкЕЅИібљБОЫЎЦНЩЯЪЙгУЁЃ
ЁОВЙГфЁПdada2КЭdeblurЯъЧщМћЃКПЦбЇЭјЁЊQIIME 2НЬГЬжЎЩњГЩЬиеїБэКЭЬиеїађСа(АИР§вЛ) - СѕЪїЧрЕФВЉЮФ (sciencenet.cn)
ЁОВЮПМЁП
QIIME2НјНзЖў_дЊЪ§ОнМАЪ§ОнЕМШыQIIME2
QIIME2НјНзШ§_гУQIIME2ЪЕЯжЖдЪ§ОнЕФжЪСППижЦ
| Ъ§ОнЯТдиЃКАйЖШЭјХЬ ЧыЪфШыЬсШЁТы (baidu.com) | УмТы1234 |
https://m.sciencenet.cn/blog-994715-1362602.html
ЩЯвЛЦЊЃКQIIME 2НЬГЬжЎЩњГЩЬиеїБэКЭЬиеїађСа(АИР§вЛ)
ЯТвЛЦЊЃКQIIME 2ађСаДІРэЧщПіЛузмЯъНт