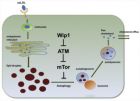
脑老化机制与认知衰退 Neural mechanisms of ageing and cognitive decline 美国哈佛医学院病理系 Nicholas A.Bishop Tao Lu Bruce A. Yankner 摘要 在过去的二十世纪,针对中青年疾病的治疗手段显著延长了人类寿命。然而,认知衰退现已成为老龄人口的重要威胁之一; 85 岁以上人群中近半数罹患阿尔茨海默病。研发针对此类疾病的治疗干预手段,需要深入阐释人脑由生理性衰老到病理性衰老的潜在演变过程。衰老生物学在模式生物( model organisms )方面的进展,以及在人脑分子和系统水平的研究,正在揭示相关机制及其潜在作用在认知衰退中的意义。 Bishop NA, Lu T, Yankner BA. Neuralmechanisms of ageing and cognitive decline. Nature, 2010, 464, 529-535. 认知衰退 ( cognitivefrailty ) 是二十一世纪最大的健康威胁之一。伴随人类预期寿命的延长,认知衰退和痴呆的发病率日渐升高;在美国,占其中大多数的阿尔茨海默病 ( AD ) ,已经影响到近半数的 85 岁以上人口 。而人口平均寿命还将增加,这一惊人数字还会上升;所以阐明衰老过程中认知衰退的机制是极为紧迫的。而老年人发生认知衰退和 AD 最重要的危险因素正是年龄本身。因此,要研究这些病症的发病机制必须理解衰老过程的分子生物学背景。 幸运的是,过去 15 年间,为理解衰老的基本分子机制,已经发现大量的证据。最引人注目的是,功能性遗传分析 ( functionalgenetic analysis ) 发现,衰老和寿命信号通路的主调节因子 ( master regulator ) 在酵母、线虫、果蝇和哺乳类具有保守性。针对这些模式生物系统的分析提示,衰老的速率不仅不是绝对固定的,而且是可塑的 ( plastic ) 和可修正的 ( open to modification ) 。类似地,哺乳类脑老化相关的认知衰退似乎也具有多样性,并有可能延缓其进展 (表 1 ) 。重要问题是,在模式生物中发现的衰老和寿命主调节因子,是否同样介导增龄性认知变化过程。而且,近期研究提示,上述相关通路可控制增龄性脑病变;那么,通过调节衰老的基本进程以改变神经变性的发病过程,这种可能性是大大增加了。 表 1 影响模式生物衰老和哺乳类脑老化的信号通路 通路 模式生物衰老效应 哺乳类脑老化效应 胰岛素 /IGF-1 信号 减弱信号可致应激耐受增强和寿命延长 减弱信号可致 AD 病变减轻;矛盾的是,增强信号可能也具有神经保护作用 TOR 信号 减弱信号可致寿命延长、自噬增强和蛋白转译减弱 对自噬和蛋白稳态的调控作用,可能调节神经变性疾病的毒性蛋白聚集 线粒体功能 严重降低可缩短寿命,而中度降低可延长寿命 衰老过程中进行性降低可促进认知衰退和病变发生;而初步研究显示,中度降低可能诱导有益通路 sirtuins 在不同背景下均可延长或缩短寿命 在不同背景下可具有神经保护作用或造成神经元损伤 热量限制 适当热量限制可延长寿命 在衰老过程中可增强对认知功能的保护 IGF-1 :胰岛素样生长因子 -1 ; TOR :雷帕霉素靶蛋白 以下两项重要的技术进步为脑老化的生物学研究展示了新的前景。微阵列 技术可实现针对人类和模式生物的全基因表达分析 ( globalgene expression analysis ) ,由此发现了衰老过程的进化保守性。与此同时,老化人脑功能性成像首次为我们展示了的大型认知网络的工作画面。目前的重要挑战,是将上述两个层次的分析相统一,以获取人脑和衰老机体更全面的信息。 本篇综述将探讨衰老过程中脑老化的基本分子机制。首先概述人脑老化大致的功能变化;再讨论衰老的保守性机制;该机制作为脑老化表现的基础,重点包括线粒体功能和氧化应激、自噬和蛋白质周转 ( protein turnover ) 、胰岛素 /IGF 信号、雷帕霉素靶蛋白 ( TOR ) 信号及 sirtuins 功能。为能够在完整机体背景下理解脑老化,本文还将讨论人脑如何与衰老过程相协调,即人脑通过保守的信号通路的作用,发挥作为主要生理感受器和环境应激源 ( environmental stressor ) 的功能。随着对脑老化分子机制和衰老机体脑功能研究的日益深入,人类将有信心应对治疗和预防认知衰退和 AD 这一挑战。 人脑老化的轨迹 伴随机体的衰老,人脑的固有结构和神经生理学发生变化,并表现不同程度的认知衰退。近期,脑功能影像学首次提供了神经活动的整体画面及其增龄性变化 (图 1 ) 。这项研究显示,各独立脑区间通过互动( interact )实现高级认知功能;而伴随衰老,相互间的协调激活效应减弱,即提示整合功能 ( integrativefunction ) 的全面衰退 。重要的是,人脑协调功能的降低与多项认知域 ( cognitivedomain ) 评分不佳相关 。除了整合功能降低,某些脑区的神经功能也更加局限化( less localized ),特别是前额皮层对应的任务执行功能 。对比发现,在执行相同任务时,年轻个体可激活更多的独立脑区,并将这些脑区更紧密地与其他脑区相整合。表现去局限化功能 ( delocalized activity ) 的老年个体认知评分,优于具有局限化认知功能的老年个体,这与认知功能去局限化可能为认知衰退代偿反应的观点相一致 。上述结果提示,人脑的高级系统生物学功能与正常衰老 (无疾病) 显著相关。 脑高级功能增龄性损害可能与部分有髓神经纤维破坏有关 (后者连接不同皮层区的神经元) 。虽然神经元缺失在正常老化的多数脑区极少存在 ,而老化神经元突触的生理变化,则可能促发神经元连接性 ( connectivity ) 和高级整合性 ( integration ) 的变化。衰老小鼠、大鼠、猴以及人类脑基因表达谱的研究均显示,突触基因表达发生显著变化 。在人类及恒河猴 ( rhesus macaque ) 前额皮层, γ- 氨基丁酸 ( GABA ) 介导的抑制性神经传递相关基因多数均随增龄而强烈下调,从而改变神经传递抑制性和兴奋性之间的潜在平衡 。这种变化可能会促发衰老个体的前额皮层神经活动增强,而这种系统层次的变化最初可能具有补偿效应,但却导致个体易受兴奋性毒性损害,易发生神经变性病变。 图 1 脑老化过程中功能性激活的变化 脑激活的功能影像学显示,伴随人脑老化,任务操作过程中发生了激活模式的变化。 a ,上:功能性磁共振成像 ( fMRI ) 显示,年轻个体的内侧前额皮层 ( mPFC ) 、后扣带回 ( pC ) 和外侧顶叶皮层 ( LP ) 同时激活,而这一瞬时的相关性活动在老年个体则相当程度地减弱 (图像经许可复制自文献 ) 。下:推测 mPFC 、 pC 与 LP 三者间的连接可能介导年轻个体的激活协调性,而衰老个体所见脑功能协调性破坏的原因,可能在于某些连接的功能下降 (图片由帕萨迪纳市加州理工学院 C.Koch 惠赠) 。 b ,正电子发射断层扫描 ( PET ) 显示,年轻个体执行记忆测试表现为右脑激活;老年个体相同测试评分不佳者也表现为右侧前额皮层 ( PFC ) 激活,而评分良好者表现为双侧激活。因此,纳入其他脑区可能会补偿初始脑区的增龄性功能下降,有助于认知功能的实现 (图像经许可复制自文献 ) 。 问题的核心是,衰老个体常见的上述功能变化,是否可以与 AD 等神经变性疾病的病变过程截然区分。 fMRI 研究显示,海马及相关皮层区的活动变化,可鉴别正常衰老与病理性衰老。正常衰老与海马下托 ( subiculum ) 及齿状回 ( dentate gyrus ) 的代谢活动降低有关,而内嗅皮层 ( entorhinal cortex ) 代谢活动降低则是 AD 的早期指征 。在组织病理学水平,神经元缺失 (起自内嗅皮层和海马 CA1 区) 并伴内侧颞叶体积减小,可鉴别正常衰老与 AD 相关认知衰退 。不过,虽然 AD 其他病理特征 (如突触缺失、淀粉样蛋白斑块和神经原纤维缠结) 与认知衰退相关联,并在 AD 中广泛存在,但亦可不同程度见于许多无认知衰退的老年个体。欲理解 AD 与正常脑老化之间的关系,尚需更多地揭示衰老过程的本质。模式生物衰老通路的发现,有助于阐明这一基础性问题。 脑部保守性衰老通路 在线虫类动物秀丽隐杆线虫 ( Caenorhabditiselegans ) 和普通果蝇 ( Drosophila melanogaster ) 的衰老过程中,以及小鼠、大鼠、黑猩猩 ( chimpanzee ) 及人类脑老化过程中,全基因表达研究显示,广泛存在极少量功能保守的基因类型,且基因表达呈现增龄性变化 (表 2 ,图 2 ) 。特别的是,多数研究显示,衰老过程伴随线粒体功能降低。并且在人类认知衰退和 AD 患者中,线粒体能量代谢相关的基因表达降低特别明显 。另一个普遍的保守性衰老特征是,应激反应通路相关基因表达的增强。小鼠、恒河猴和人类老化皮层的转录谱 ( transcriptional profiling ) 研究显示,载脂蛋白 D 基因的增龄性上调是最具保守性的变化 。载脂蛋白 D 表达可延长果蝇寿命;该蛋白具有脂质抗氧化剂的功能,能够耐受氧化应激 。而且, AD 患者脑内载脂蛋白 D 表达增加 。总之,衰老过程中应激耐受的保守性机制亦可作用于脑部,保护其免受神经变性病变的侵袭。 尽管有确凿证据证实,某些衰老通路和基因表达特征具有保守性;然而最近一项研究直接比较小鼠、恒河猴和人类脑内的基因表达,显示在衰老过程中呈现出重要的进化趋异性 ( evolutionary divergence ) 。发现有 150 种以上的基因表达在三物种间均呈现增龄性变化;但这些基因在三物种间的表达方向并不一致。占相当比例的衰老调节基因 (据预测多数参与神经元功能) ,在小鼠为增龄性上调,而在人类为增龄性下调。以上提示,啮齿类和人类间的进化趋异性,导致人类衰老过程中许多基因表现为协同性抑制 ( coordinate repression ) 而非激活。采用生化和体视学 ( stereology ) 方法,针对老化人脑皮层的神经元计数研究提示,基因表达的变化未必能反映神经元的缺失 。但这种情形可能导致 AD 等神经变性疾病具有显著的人类特异性,所以应意识到评估上述基因表达变化的重要性。 图 2 脑老化过程中基因调节的进化性改变 灵长类基因表达具有广泛的增龄性调节变化。人类和恒河猴皮层增龄性基因变化以下调为主 (粉红) ,相对照的小鼠皮层中多数年龄调节基因被上调 (绿) 。这种程度的皮层基因抑制现象未见于非神经元的其他人体组织,包括外周血单核细胞 (作者未发表资料) 、肌肉 和肾脏 。 表 2 脑老化过程中基因表达的进化保守性 基因类型 人类 (脑) 恒河猴 (脑) 大鼠 (脑) 小鼠 (脑) 果蝇 (机体) 线虫 (机体) 应激反应 ↑ ↑ ↑ ↑ ↑ ↑ 线粒体 ↓ ↓ ↓ ↓↑ ↓ ↓ 神经可塑性 / 突触功能 ↓ ↓ ↓ ↓ — — 抑制性中间神经元功能 ↓ ↓ — — — — 泛素 - 蛋白酶体通路 ↓ ↓ — ↓ — — 免疫 / 炎症反应 ↑ — ↑ ↑ ↑ — 金属离子稳态 ↑ — ↑ — — — 髓鞘相关蛋白 ↑ — ↑ — — — 神经胶质基因 ↑ — ↑ — — — 应激反应和线粒体功能等某些相关基因类型,在衰老过程中可呈现保守性变化;而抑制性中间神经元功能等基因类型,则表现为灵长类特异性变化。向上箭头示表达随龄增加,向下示表达随龄减少,横线示表达随龄无变化。在衰老小鼠脑内,不同线粒体基因可为上调或下调 。 线粒体功能 从线虫到人类诸多物种的基因表达研究提示,线粒体基因表达降低是具有强烈保守性的衰老特征 。特别是限定于脑老化进行的分析显示,大鼠、恒河猴和人类线粒体基因表达均呈现进行性下降 。在所有检测物种中发现,线粒体功能可能为衰老进程的重要调节因素;在不同背景下,线粒体功能可对寿命产生正性或负性效应。 减弱线粒体功能可能会损害健康、缩短寿命;且确有发生于非脊椎动物和哺乳类的实例。严重削弱线粒体功能可显著缩短线虫寿命 。线粒体 DNA ( mDNA ) 突变率升高的基因工程小鼠表现为电子传递链功能降低、衰老体征加速和寿命缩短 。与之相反,加强线粒体功能显示可延长寿命。抗氧化剂过氧化氢酶的线粒体靶向性过表达 ( targeted overexpression ) 可充分延长小鼠寿命 。而且,人工提高酵母的线粒体呼吸速率,也可充分延长其复制性寿命 ( replicativelifespan ) 。虽然上述实验模型寿命延长的确切机制尚不完全清楚,但有一种假说认为,高效的电子传递链功能可减少损害性活性氧 ( ROS ) 的生成和释放。 脑和肌肉特别易于受到线粒体功能缺陷的影响。人类线粒体脑肌病 ( mitochondrialencephalomyopathies ) 为 mDNA 删除或突变所致的遗传性疾病。这种线粒体缺陷可导致各种神经和肌肉相关损害,取决于各细胞中线粒体受损数量的不同 。重要的是,线粒体受损数量还可调节临床症状的发生年龄。所以,这一发现提示,线粒体功能的正常降低,也可能促进神经元和肌细胞的增龄性功能损害。支持此观点的证据来自果蝇研究:作为哺乳类脑内特异性线粒体解偶联蛋白 ( mitochondrial uncoupling protein ) 在果蝇的直系同源物 ( orthologue ) , UCP5 可通过发挥神经元特异性功能以调节果蝇代谢和寿命 。在人脑,线粒体功能降低可能会选择性损伤生物能 ( bioenergy ) 需求量较大的细胞,如大锥体神经元 ( large pyramidal neurons ) 变性则可致 AD 。因此,线粒体功能降低可促进脑老化,并增加神经元对增龄性病变的易感性。 出乎意料的是,在某些情况下,降低线粒体功能亦可延长寿命。一项早期的研究提示来自于对 clk-1 突变线虫的分析 。 CLK-1 为合成泛醌 ( ubiquinone ) 所必需,后者在线粒体呼吸中具有重要作用,故而 clk-1 突变线虫线粒体呼吸速率降低。并且,这些线虫的寿命延长,发育和行为的进度普遍延迟。随后通过 RNA 干扰 ( RNA interference , RNAi ) 筛选发现,降低电子传递链相关的基因功能能够延长寿命 。这种效应似乎具有严格的剂量依赖性,因为电子传递链功能中度降低才能延长寿命,而更进一步降低功能则致寿命缩短 。新近证据提示,上述寿命延长效应可能由某种核转录反应所介导;该反应针对线粒体损害而产生,称为逆行反应 ( retrograderesponse ) ,涉及到诱导氧化应激耐受和有害异物解毒基因 ( xenobioticdetoxification gene ) 。在线虫,特异性减少成虫神经元的电子传递链成分表达,可充分延长其寿命 。这种现象也可见于哺乳类,因为 Coq7 杂合突变小鼠同样长寿 ( Coq7 为 clk-1 在小鼠的直系同源基因) 。而且,表现为细胞色素 c 氧化酶复合体 (为电子传递链成分) 活性降低的某种小鼠模型,同样实现寿命延长 。有趣的是,这种小鼠还具有保护功能以对抗脑内神经元兴奋性毒性的侵袭。在此背景下,虽然信号通路介导的寿命延长效应尚未完全阐明,但有一种可能性认为,中等程度增加的 ROS 可作为信号分子激活生存通路 ( survival pathway ) ,以促进寿命延长。 中度降低线粒体功能可激活长寿通路,此发现为下列现象提供了可能的有趣解释,即脑老化的初始阶段线粒体基因表达减弱,可能属于主动性代偿机制 (增强应激耐受) 的一部分。这种代偿反应可有效耐受短暂应激,但衰老相关的持续应激则会进一步降低线粒体功能,导致形成线粒体功能降低并自我强化的恶性循环 (图 3 ) 图 3 调节机体衰老和脑老化的保守通路 所示为线粒体功能、氧化应激、自噬、蛋白质稳态、 TOR 信号、胰岛素 /IGF-1 信号 ( IIS ) 、热量限制 ( CR ) 和 sirtuins 的相关作用机制。线粒体正常代谢产生适量的 ROS ,可诱导应激耐受通路以清除 ROS 并修复损伤。然而,线粒体进行性损伤却导致 ROS 病理性聚集,后者可促进线粒体的进一步损伤。受损线粒体可通过自噬途径清除; CR 可促进自噬,而 TOR 信号则抑制自噬。 CR 改善线粒体的整体功能,部分通过促进线粒体的生物合成 ( biogenesis ) 和减少 ROS 产生而实现 。 ROS 亦可损伤诸如 DNA 、蛋白质等其它关键分子。未修复的受损 DNA 会出现表观遗传学 ( epigenetics ) 变化和基因沉默 ( genesilencing ) ,并通过诱导核编码线粒体基因表达而加重线粒体损伤。 ROS 也可修饰蛋白质,导致蛋白质去折叠 ( unfolding ) 和聚集。修饰的蛋白质可通过泛素 - 蛋白酶体通路 ( ubiquitin–proteasomepathway ) 等降解通路清除。清除不足则可致毒性蛋白的聚集。蛋白清除和聚集形成的动力学过程受到 IIS 通路、 SIRT1 和 CR 的调节。而 TOR 信号和 CR 也可通过 mRNA 转译途径,调节毒性受损蛋白聚集体的修饰。 氧化应激与表观遗传学变化 多物种的证据提示,进行性氧化应激是增龄性功能下降的保守性核心机制 。针对衰老线虫和果蝇整体以及小鼠、大鼠、黑猩猩和人类老化脑进行的基因表达研究显示,这六个物种均表现为氧化应激反应基因的增龄性上调 。而且,在老化人脑的前额皮层,介导氧化应激反应和 DNA 损伤修复的基因, 涵盖了上调基因的绝大多数 。膳食抗氧化剂 ( dietaryantioxidant ) 可抑制小鼠脑内多种增龄性基因表达 ,并减轻衰老大鼠的认知衰退,预防脑内氧化损伤 。 在老化人脑,基因启动子的特异性氧化损伤可致基因沉默 。原因可能是,作为不可替代的有丝分裂后细胞,神经元针对未修复 DNA 作出的反应,倾向于通过受影响基因组片段的表达沉默,而非通过凋亡途径而实现。表达沉默的机制可能为表观遗传学效应;确切说法是,将神经元转变到更具抑制性的转录状态 。近期研究显示, DNA 损伤可诱导基因表达的变化,保守的寿命调节基因 SIRT1 (见下) 可部分介导组蛋白的修饰模式;提示存在可能的机制将 DNA 损伤、表观遗传学状态和衰老三者联系起来 。与此观点一致, SIRT1 在 酵母的直系同源基因 SIR2 ,早已被认定为寿命、 DNA 损伤修复和表观遗传基因沉默的调节因子 。而且,有多种染色质重组因子 ( chromatin-remodelling factor ) 可调节线虫寿命 。上述例证强烈支持,表观基因组动力学 ( epigenomedynamics ) 在衰老过程中具有保守性作用。 近期研究显示,控制神经元基因表达的表观遗传学变化,是影响突触可塑性和记忆过程重要的深层次因素。诱导表达 p25 的小鼠模型可引起神经变性。成年小鼠脑内 p25 短暂表达,可致海马一定程度的神经变性、突触缺失,并伴记忆丧失 。环境强化 ( environmentalenrichment ) 可促进遗忘记忆的恢复,并可增强突触可塑性,诱导组蛋白乙酰化标记的激活。值得注意的是,采用具有药理作用的组蛋白去乙酰酶抑制剂治疗,可模拟上述环境强化效应,促进神经元可塑性和记忆功能的恢复。这些发现凸显了表观遗传学变化在神经变性相关记忆丧失过程中的作用。另外,上述研究还提示,记忆储存区 ( memory storage ) 的丧失与进入该区域的神经通路丧失并无关联。假如脑老化伴记忆丧失和突触连接性降低,但不伴神经元缺失,则原因很可能在于,针对已储存记忆的获取功能丧失,造成了增龄性记忆损害。若此,有望通过药物干预影响表观遗传学状态,进而减轻衰老和神经变性相关的某些认知损害。 自噬与蛋白质周转 模式生物研究提示,自噬是衰老过程决定性的调节因素。在线虫,降低胰岛素样信号 和饮食限制 可致自噬增强,自噬增强则为寿命延长所必须。在果蝇,单独增强神经元自噬即可充分延长寿命 ;降低自噬则致寿命缩短,发生神经变性 。降低小鼠神经系统的基础自噬亦可引起类似的神经变性 。在自噬缺陷果蝇和小鼠,神经变性伴随泛素化蛋白聚集体的堆积,类似于人类亨廷顿病 ( HD ) 和 AD 等神经变性疾病所见。与此一致, BECN1 (自噬关键调节因子之一) 表达与突变亨廷顿蛋白 ( huntingtin ) 聚集体的有效清除直接相关 。人脑 BECN1 及其他自噬通路关键基因的表达随衰老而降低 (作者未发表资料) ,潜在增加神经元针对蛋白聚集体毒性效应的易感性。 激酶 TOR 信号通路是蛋白质稳态的核心调节因子,其功能为抑制自噬和 mRNA 转译。研究显示,降低 TOR 信号可延长酵母、线虫和果蝇寿命 。并且,近期研究显示,即使在小鼠生命晚期应用 TOR 抑制剂雷帕霉素,亦可延长其寿命 。目前的认知程度还无法明确,神经元中何种 TOR 信号可促进保守的寿命延长效应;但有证据显示, TOR 信号和自噬可对病理性蛋白聚集体 (与增龄性神经变性疾病相关) 产生显著效应。例如,雷帕霉素应用于小鼠和果蝇的 HD 和 tau 蛋白病 ( tauopathy ) 模型,均显示可通过增强自噬以减少毒性聚集体形成和疾病进展 。上述结果提示, TOR 通路或可调节神经变性疾病病理性蛋白聚集。 泛素化蛋白聚集体的堆积可发生于正常老化人脑,而在 AD 和 tau 蛋白病等神经变性疾病,则达到病理性水平。近期研究显示,缓慢降低小鼠脑内蛋白酶体活性,可升高数种蛋白水平,其中某些蛋白的表达变化在 AD 脑内亦有显示 。而且,这些小鼠存在空间记忆缺损,与蛋白酶体功能缺陷在 AD 相关认知衰退中导致的效应相一致。泛素化蛋白聚集体在其他衰老组织中并不常见,这可能反映出有丝分裂后神经元具有与众不同的长寿效应 (处于代谢激活状态的该神经元可存活于人类整个生命期) 。 胰岛素 /IGF 信号 在线虫、果蝇和哺乳类,降低胰岛素 /IGF-1 信号 ( IIS ) 通路可实现寿命延长,这可归因于强烈的保守性机制 。两条 IIS 通路基因的多态性、 IGF-1 受体及下游 FOXO3 转录因子,均与人类长寿和健康衰老 ( healthyageing ) 相关 。重要的是,在多种模型系统中特异性降低神经系统 IIS ,均可充分延长寿命 。并且,果蝇脑内存在特异性产生胰岛素样肽的神经内分泌细胞,消融该细胞亦可充分延长果蝇寿命 。 在哺乳类,胰岛素和 IGF-1 具有神经营养作用,可通过抑制凋亡促进神经元存活 。胰岛素和 IGF-1 也可增强人类和动物模型的学习和记忆能力 。比较发现,通过特异性敲除神经元 Irs2 ( 胰岛素受体底物) ,可降低 IIS 并充分延长小鼠寿命 。这即是胰岛素和 IGF-1 信号的两面性,其既具有神经保护效应,对机体寿命又具有明显的毒性效应。有趣的是,其针对寿命的效应与针对神经变性病变的效应相平行。在线虫,降低胰岛素信号可减轻 β- 淀粉样蛋白聚集和细胞毒性 。类似地,在小鼠 AD 模型,敲除 Irs2 ,或 IGF-1 受体基因可减轻认知损害和神经变性病变,并降低早衰死亡率 。有证据显示, AD 患者中 IGF 信号通路成分的表达降低 。但尚不清楚这种现象是主动的神经保护反应,抑或是神经变性过程的继发表现。 热量限制和 sirtuins 热量限制 (减少热量摄入并避免营养不良) 是已知唯一可稳定延长多种物种寿命的干预手段 。近期,这一现象在灵长类亦得以延续:一项针对恒河猴热量限制的长期实验显示其寿命得到延长 。在改善脑功能和削弱增龄性病变易感性方面,热量限制亦具有广泛的有益功效。热量限制可阻止小鼠脑内多种增龄性基因表达变化 ,减轻恒河猴增龄性脑萎缩 ;并对于啮齿类 和人类 增龄性学习和记忆损伤,也具有显著的有益功效。出乎意料的是,针对健康衰老人群进行三个月的热量限制,其词语记忆力即可提高约 20% 。还有证据显示,热量限制或可使机体具有耐受 AD 病变的能力:研究表明,针对转基因 AD 小鼠模型,热量限制可降低 β- 淀粉样蛋白沉积,改善学习和记忆能力。 近期研究已经开始揭示热量限制有益功效的分子机制 。针对热量限制, NAD + 依赖性去乙酰酶 SIR2 是最早发现的相关基因之一 (最初发现于酵母模型) 。随后发现, sirtuins ( Sir2 同系物) 可介导热量限制在果蝇 和哺乳类 的部分效应。但值得重视的是, sirtuins 在热量限制中的潜在功效是有争议的,并且是背景依赖的 (见前篇) ;在不同遗传背景的酵母 和不同类型的小鼠组织 ,可显示截然不同的效应。类似地, SIRT1 在脑内亦可产生有益或有害效应。 SIRT1 针对脑老化显示有益效应的证据如下,在缓慢型华勒变性 ( Wallerian degeneration ) 突变小鼠发现,增强 SIRT1 活性可防止损伤后轴突变性 。 SIRT1 还可保护培养细胞对抗 β- 淀粉样蛋白毒性,保护 p25/CDK5 小鼠模型避免神经变性;这两方面的作用涵盖了 AD 和 tau 蛋白病的特征 。与之相反, SIRT1 也可能具有某些有害效应。例如, Sirt1 敲除衰老小鼠脑内显示氧化应激减弱,神经保护增强,但其寿命缩短 。尚需深入研究以阐明 sirtuins 在脑老化过程中的作用,及其针对神经变性疾病潜在的治疗功效。 衰老机体脑调节的潜在机制 越来越多的证据显示,通过协调非神经元组织的生理效应,神经系统可作为衰老的调节中枢。在线虫,多种类型的突变在破坏感觉神经元功能的同时,又延长了机体寿命 。而且,特异性消融神经元亦可延长线虫 和果蝇 寿命。神经系统作为调节中枢的一个著名实例是,线虫的一对神经元 ( ASI 神经元) 在热量限制条件下可介导寿命延长 。寿命延长有赖于转录因子 SKN-1 在 ASI 神经元中的活性,已知该转录因子的功能是激活细胞以防御氧化应激和有害异物应激。有趣的是, ASI 神经元调节线虫能量代谢,可能类似于哺乳类下丘脑的功能。 下丘脑是代谢调节和能量利用的中枢;通过激素信号,下丘脑协调整个机体的生理反应。在小鼠,降低来自下丘脑 - 垂体轴的生长激素信号可延长寿命 。并且,针对小鼠下丘脑特定细胞过表达线粒体解偶联蛋白 UCP2 ,亦可充分延长其寿命 。上述发现证实,在调节机体衰老方面,下丘脑激素信号发挥关键性作用。 图 4 脑在机体衰老中具有的潜在调节作用 脑老化可能通过激素反馈环路与其他器官系统老化相协调。图中左侧示意,糖皮质激素信号如何促进脑老化。应激源诱导下丘脑产生促肾上腺皮质激素释放激素 ( CRH ) ,导致肾上腺释放糖皮质激素。随后,海马通过糖皮质激素受体 ( GR ) 感受糖皮质激素水平,并反馈性抑制糖皮质激素通过下丘脑途径释放。衰老过程中,缓慢升高的糖皮质激素可能会损害海马功能,削弱海马抑制糖皮质激素释放的功能,造成激素介导的海马功能衰退,这是一个潜在的自我强化过程。图中右侧示意 (与左侧通路类似) ,激素反馈环路如何成为促进脑区其他系统功能衰退的基本机制。虚线箭头示意该效应为间接性和 / 或机制未完全阐明。 下丘脑调节激素通路的情形,也表现在脑老化过程中出现的认知衰退上。下丘脑协调应激反应,部分通过调节外周糖皮质激素分泌实现 。而在另一脑区的海马亦可直接感知糖皮质激素,并可抑制下丘脑发出的释放更多糖皮质激素的刺激,从而形成负反馈环路 (图 4 ) 。然而,慢性或严重应激导致过量糖皮质激素产生可能损害海马神经元功能,增加机体对神经变性的易感性 ,并潜在破坏连接海马和下丘脑的调节环路。并且,作为 AD 患者主要表现的海马神经变性,也可破坏海马 - 下丘脑对全身生理功能的调节 。探索其他的脑 - 机体环路可能进一步整合脑的高级功能与机体生理机能。因此,脑部重要的变性病变可促使机体对糖皮质激素及其他重要激素 (如 IGF-1 ) 的调节作用衰退。而这种全身性变化可能会进一步加重神经变性,从而形成生理机能恶化与神经功能衰退之间进行性加重的恶性循环 (图 4 ) 。 未来研究方向 神经变性和认知衰退最重要的危险因素是脑老化。调节机体衰老的保守性通路和机制,如胰岛素 /IGF-1 信号和线粒体功能,也可调节神经病变和认知衰退 (在 AD 等神经变性疾病的小鼠、果蝇、线虫模型中均有显示) 。然而,在人类神经变性疾病的发生和进展过程中,这些保守性通路所发挥的作用仍不明确。这项基础性问题的解决,有待于未来针对这些通路开展临床干预,以确定这些通路在正常增龄性认知衰退和病理性神经变性中的作用。 脑在衰老过程中扮演 “ 生理变化整合中枢 ” ( centralintegrator of physiological changes ) 的角色,而相关的研究探索才刚刚开始。近期研究提示,伴随衰老过程,脑系高级功能的效率降低,脑区间联络出现不同程度中断 (在年轻人则正常) 。这可能部分反映,基因表达变化影响到突触功能、轴突完整性和髓鞘形成。引人深思的问题是,是否脑内功能性联络中断会导致脑 - 机体反馈环 (涉及重要激素和自主神经系统) 的破坏。上述整合功能的缺失可能会促进增龄性生理变化 (如高血压、胰岛素抵抗) ,增加个体对增龄性脑病变的易感性。在未来研究中,深入到上述功能连接层次进行探索,将会是激动人心的。 参考文献 1. Hebert, L. E.,Scherr, P. A., Bienias, J. L., Bennett, D. A. Evans, D. A. Alzheimer diseasein the US population: prevalence estimates using the 2000 census. Arch. Neurol. 60, 1119–1122 (2003). 2. Andrews-Hanna,J. R. et al . Disruption of large-scale brain systems in advanced aging. Neuron 56, 924–935 (2007). 本文报道老化脑的不同脑区间协同活动的稳定性降低,提示认知衰退与系统水平的整合功能破坏有关。 3. Cabeza, R. Hemisphericasymmetry reduction in older adults: the HAROLD model. Psychol. Aging 17, 85–100 (2002). 本文显示在人脑前额皮层存在扩散性激活 ( spreadingactivation ) ,可由一侧半球分布到双侧半球,这可能为脑部保持功能以对抗增龄性神经变性病变的代偿性机制。 4. Park, D. C. Reuter-Lorenz, P. The adaptive brain: aging and neurocognitive scaffolding. Annu.Rev. Psychol. 60, 173–196 (2009). 5. Cabeza, R., Anderson,N. D., Locantore, J. K. McIntosh, A. R. Aging gracefully: compensatory brainactivity in high-performing older adults. Neuroimage 17, 1394–1402 (2002). 6. Yankner, B. A.,Lu, T. Loerch, P. The aging brain. Annu. Rev. Pathol. 3, 41–66 (2008). 7. Lu, T. etal . Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891 (2004). 本文显示转录信号的显著变化是老化人脑皮层的特征,该变化包括介导突触可塑性的基因表达降低,而这与这些基因启动子发生增龄性 DNA 损害有关。 8. Lee, C. K., Weindruch,R. Prolla, T. A. Gene-expression profile of the ageing brain in mice. NatureGenet. 25, 294–297 (2000). 9. Jiang, C. H.,Tsien, J. Z., Schultz, P. G. Hu, Y. The effects of aging on gene expressionin the hypothalamus and cortex of mice. Proc. Natl Acad. Sci. USA 98, 1930–1934 (2001). 10. Blalock, E. M. et al . Gene microarrays in hippocampal aging: statistical profiling identifiesnovel processes correlated with cognitive impairment. J. Neurosci. 23, 3807–3819 (2003). 11. Fraser, H. B.,Khaitovich, P., Plotkin, J. B., Paabo, S. Eisen, M. B. Aging and gene expressionin the primate brain. PLoS Biol. 3,e274 (2005). 12. Erraji-Benchekroun,L. et al . Molecular aging in human prefrontal cortex is selective and continuousthroughout adult life. Biol. Psychiatry 57, 549–558 (2005). 13. Loerch, P. M. et al . Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 3, e3329 (2008). 14. Small, S. A.,Tsai, W. Y., DeLaPaz, R., Mayeux, R. Stern, Y. Imaging hippocampal functionacross the human life span: is memory decline normal or not? Ann. Neurol. 51, 290–295 (2002). 15. West, M. J.,Coleman, P. D., Flood, D. G. Troncoso, J. C. Differences in the pattern ofhippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 344, 769–772 (1994). 16. Price, J. L. et al . Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimerdisease. Arch. Neurol. 58,1395–1402 (2001). 17. Gomez-Isla, T. et al . Profound loss of layer II entorhinal cortex neurons occurs in verymild Alzheimer's disease. J. Neurosci. 16, 4491–4500 (1996). 18. Rodrigue, K.M. Raz, N. Shrinkage of the entorhinal cortex over five years predicts memoryperformance in healthy adults. J. Neurosci. 24, 956–963 (2004). 19. Zahn, J. M. etal . AGEMAP: a gene expression database for aging in mice. PLoS Genet. 3, e201 (2007). 20. Blalock, E. M. et al . Incipient Alzheimer's disease: microarray correlation analyses revealmajor transcriptional and tumor suppressor responses. Proc. Natl Acad. Sci. USA 101, 2173–2178 (2004). 21. Liang, W. S. et al . Alzheimer's disease is associated with reduced expression of energymetabolism genes in posterior cingulate neurons. Proc. Natl Acad. Sci. USA 105, 4441–4446 (2008). 22. Miller, J. A.,Oldham, M. C. Geschwind, D. H. A systems level analysis of transcriptionalchanges in Alzheimer's disease and normal aging. J. Neurosci. 28, 1410–1420 (2008). 23. Walker, D. W.,Muffat, J., Rundel, C. Benzer, S. Overexpression of a Drosophila homologof apolipoprotein D leads to increased stress resistance and extended lifespan. Curr. Biol. 16, 674–679 (2006). 24. Sanchez, D. etal . Loss of glial lazarillo, a homolog of apolipoprotein D, reduces lifespanand stress resistance in Drosophila . Curr. Biol. 16, 680–686 (2006). 25. Kalman, J., McConathy,W., Araoz, C., Kasa, P. Lacko, A. G. Apolipoprotein D in the aging brain andin Alzheimer's dementia. Neurol. Res. 22, 330–336 (2000). 26. Sedensky, M.M. Morgan, P. G. Mitochondrial respiration and reactive oxygen species inmitochondrial aging mutants. Exp. Gerontol. 41, 237–245 (2006). 27. Rea, S. L., Ventura,N. Johnson, T. E. Relationship between mitochondrial electron transport chaindysfunction, development, and life extension in Caenorhabditis elegans . PLoSBiol. 5, e259 (2007). 28. Trifunovic, A. et al . Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004). 29. Kujoth, G. C. et al . Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalianaging. Science 309, 481–484(2005). 30. Schriner, S.E. et al . Extension of murine life span by overexpression of catalase targetedto mitochondria. Science 308,1909–1911 (2005). 31. Lin, S. J. etal . Calorie restriction extends Saccharomyces cerevisiae lifespan byincreasing respiration. Nature 418,344–348 (2002). 32. Wallace, D. C. et al . Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological,and biochemical characterization of a mitochondrial DNA disease. Cell 55, 601–610 (1988). 33. Holt, I. J.,Harding, A. E. Morgan-Hughes, J. A. Deletions of muscle mitochondrial DNAin patients with mitochondrial myopathies. Nature 331, 717–719 (1988). 34. Sanchez-Blanco,A., Fridell, Y. W. Helfand, S. L. Involvement of Drosophila uncouplingprotein 5 in metabolism and aging. Genetics 172, 1699–1710 (2006). 35. Branicky, R.,Bénard, C. Hekimi, S. clk-1 , mitochondria, and physiological rates. BioEssays 22, 48–56 (2000). 36. Lee, S. S. etal . A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nature Genet. 33, 40–48 (2003). 37. Dillin, A. etal . Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 (2002). 本文报道,通过 RNAi 介导敲除多种电子传递链成分,首次从系统水平显示,电子传递链可对后生动物 ( metazoan ,多细胞动物) 寿命发挥调节作用。 38. Cristina, D.,Cary, M., Lunceford, A., Clarke, C. Kenyon, C. A. Regulated response to impairedrespiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000450 (2009). 39. Copeland, J.M. et al . Extension of Drosophila life span by RNAi of the mitochondrialrespiratory chain. Curr. Biol. 19,1591–1598 (2009). 40. Liu, X. etal . Evolutionary conservation of the clk-1 -dependent mechanism of longevity:loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434 (2005). 41. Dell'agnello,C. et al . Increased longevity and refractoriness to Ca 2+ -dependentneurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444 (2007). 42. Muller, F. L.,Lustgarten, M. S., Jang, Y., Richardson, A. Van Remmen, H. Trends in oxidativeaging theories. Free Radic. Biol. Med. 43, 477–503 (2007). 43. Park, S.-K. etal . Gene expression profiling of aging in multiple mouse strains: identificationof aging biomarkers and impact of dietary antioxidants. Aging Cell 8, 484–495 (2009). 44. Liu, J. etal . Memory loss in old rats is associated with brain mitochondrial decay andRNA/DNA oxidation: partial reversal by feeding acetyl- L -carnitine and/or R -α-lipoic acid. Proc. Natl Acad.Sci. USA 99, 2356–2361 (2002). 本文报道,老年大鼠的记忆丧失能够实现逆转,具体是通过膳食性线粒体底物和抗氧化剂以恢复线粒体功能。 45. Oberdoerffer,P. et al . SIRT1 redistribution on chromatin promotes genomic stability butalters gene expression during aging. Cell 135, 907–918 (2008). 46. O'Hagan, H. M.,Mohammad, H. P. Baylin, S. B. Double strand breaks can initiate gene silencingand SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 4, e1000155 (2008). 47. Guarente, L.Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14, 1021–1026 (2000). 48. Curran, S. P. Ruvkun, G. Lifespan regulation by evolutionarily conserved genes essentialfor viability. PLoS Genet. 3,e56 (2007). 49. Fischer, A.,Sananbenesi, F., Wang, X., Dobbin, M. Tsai, L. H. Recovery of learning andmemory is associated with chromatin remodelling. Nature 447, 178–182 (2007). 本文显示,某种神经变性模型小鼠的记忆丧失与染色质结构变化相关,可通过组蛋白去乙酰酶抑制剂部分逆转之。 50. Melendez, A. et al . Autophagy genes are essential for dauer development and life-spanextension in C. elegans . Science 301, 1387–1391 (2003). 51. Hansen, M. etal . A role for autophagy in the extension of lifespan by dietary restrictionin C. elegans . PLoS Genet. 4, e24 (2008). 52. Simonsen, A. et al . Promoting basal levels of autophagy in the nervous system enhanceslongevity and oxidant resistance in adult Drosophila . Autophagy 4, 176–184 (2008). 53. Juhasz, G., Erdi,B., Sass, M. Neufeld, T. P. Atg7-dependent autophagy promotes neuronal health,stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila . Genes Dev. 21, 3061–3066 (2007). 54. Hara, T. etal . Suppression of basal autophagy in neural cells causes neurodegenerativedisease in mice. Nature 441,885–889 (2006). 55. Komatsu, M. etal . Loss of autophagy in the central nervous system causes neurodegenerationin mice. Nature 441, 880–884(2006). 文献 54 , 55 报道了关键性自噬调节基因缺陷模型小鼠,显示自噬可发挥对抗增龄性神经变性和蛋白聚集的重要作用。 56. Shibata, M. etal . Regulation of intracellular accumulation of mutant Huntingtin by Beclin1. J. Biol. Chem. 281, 14474–14485(2006). 57. Schieke, S. M. Finkel, T. Mitochondrial signaling, TOR, and life span. Biol. Chem. 387, 1357–1361 (2006). 58. Harrison, D.E. et al . Rapamycin fed late in life extends lifespan in genetically heterogeneousmice. Nature 460, 392–395(2009). 59. Ravikumar, B. et al . Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamineexpansions in fly and mouse models of Huntington disease. Nature Genet. 36, 585–595 (2004). 60. Fischer, D. F. et al . Long-term proteasome dysfunction in the mouse brain by expressionof aberrant ubiquitin. Neurobiol. Aging 30, 847–863 (2009). 61. Broughton, S. Partridge, L. Insulin/IGF-like signalling, the central nervous system andaging. Biochem. J. 418, 1–12(2009). 62. Flachsbart, F. et al . Association of FOXO3A variation with human longevity confirmedin German centenarians. Proc. Natl Acad. Sci. USA 106, 2700–2705 (2009). 63. Suh, Y. etal . Functionally significant insulin-like growth factor I receptor mutationsin centenarians. Proc. Natl Acad. Sci. USA 105, 3438–3442 (2008). 64. Willcox, B. J. et al . FOXO3A genotype is strongly associated with human longevity. Proc. Natl Acad. Sci. USA 105,13987–13992 (2008). 65. Libina, N., Berman,J. R. Kenyon, C. Tissue-specific activities of C. elegans DAF-16 inthe regulation of lifespan. Cell 115,489–502 (2003). 66. Iser, W. B.,Gami, M. S. Wolkow, C. A. Insulin signaling in Caenorhabditis elegans regulates both endocrine-like and cell-autonomous outputs. Dev. Biol. 303, 434–447 (2007). 67. Broughton, S.J. et al . Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl Acad. Sci. USA 102, 3105–3110 (2005). 68. van der Heide,L. P., Ramakers, G. M. J. Smidt, M. P. Insulin signaling in the central nervoussystem: learning to survive. Prog. Neurobiol. 79, 205–221 (2006). 69. Taguchi, A.,Wartschow, L. M. White, M. F. Brain IRS2 signaling coordinates life span andnutrient homeostasis. Science 317,369–372 (2007). 70. Cohen, E., Bieschke,J., Perciavalle, R. M., Kelly, J. W. Dillin, A. Opposing activities protectagainst age-onset proteotoxicity. Science 313, 1604–1610 (2006). 71. Freude, S. etal . Neuronal IGF-1 resistance reduces Aβ accumulation and protects against prematuredeath in a model of Alzheimer's disease. FASEB J. 23, 3315–3324 (2009). 72. Cohen, E. etal . Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169 (2009). 73. Moloney, A. M. et al . Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer'sdisease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol.Aging 31, 224–243 (2008). 74. Haigis, M. C. Guarente, L. P. Mammalian sirtuins — emerging roles in physiology, aging,and calorie restriction. Genes Dev. 20, 2913–2921 (2006). 75. Colman, R. J. et al . Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204 (2009). 本文报道,热量限制可延长灵长类寿命,延缓增龄性疾病的发生,并减轻增龄性脑萎缩。 76. Ingram, D. K.,Weindruch, R., Spangler, E. L., Freeman, J. R. Walford, R. L. Dietary restrictionbenefits learning and motor performance of aged mice. J. Gerontol. 42, 78–81 (1987). 77. Stewart, J.,Mitchell, J. Kalant, N. The effects of life-long food restriction on spatialmemory in young and aged Fischer 344 rats measured in the eight-arm radial and theMorris water mazes. Neurobiol. Aging 10, 669–675 (1989). 78. Witte, A. V.,Fobker, M., Gellner, R., Knecht, S. Flöel, A. Caloric restriction improvesmemory in elderly humans. Proc. Natl Acad. Sci. USA 106, 1255–1260 (2009). 79. Halagappa, V.K. et al . Intermittent fasting and caloric restriction ameliorate age-relatedbehavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 26, 212–220(2007). 80. Mair, W. Dillin, A. Aging and survival: the genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 77, 727–754(2008). 81. Lin, S. J., Defossez,P. A. Guarente, L. Requirement of NAD and SIR2 for life-span extensionby calorie restriction in Saccharomyces cerevisiae . Science 289, 2126–2128 (2000). 82. Rogina, B. Helfand, S. L. Sir2 mediates longevity in the fly through a pathway related to calorierestriction. Proc. Natl Acad. Sci. USA 101, 15998–16003 (2004). 83. Boily, G. etal . SirT1 regulates energy metabolism and response to caloric restriction inmice. PLoS ONE 3, e1759 (2008). 84. Chen, D., Steele,A. D., Lindquist, S. Guarente, L. Increase in activity during calorie restrictionrequires Sirt1. Science 310,1641 (2005). 85. Kaeberlein, M.,Kirkland, K. T., Fields, S. Kennedy, B. K. Sir2-independent life span extensionby calorie restriction in yeast. PLoS. Biol. 2, e296 (2004). 86. Chen, D. etal . Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 (2008). 87. Araki, T., Sasaki,Y. Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation preventaxonal degeneration. Science 305,1010–1013 (2004). 88. Kim, D. etal . SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer'sdisease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 (2007). 89. Li, Y., Xu, W.,McBurney, M. W. Longo, V. D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2signaling and protects neurons. Cell Metab. 8, 38–48 (2008). 90. Apfeld, J. Kenyon, C. Regulation of lifespan by sensory perception in Caenorhabditis elegans . Nature 402, 804–809 (1999). 91. Alcedo, J. Kenyon, C. Regulation of C. elegans longevity by specific gustatory and olfactoryneurons. Neuron 41, 45–55(2004). 92. Bishop, N. A. Guarente, L. Two neurons mediate diet-restriction-induced longevity in C.elegans . Nature 447, 545–549(2007). 本文显示,线虫的一对神经元可协调机体针对热量限制的反应,从而发挥关键作用;故认为线虫神经系统在这种延长寿命模式中居于核心地位。 93. Flurkey, K.,Papaconstantinou, J., Miller, R. A. Harrison, D. E. Lifespan extension anddelayed immune and collagen aging in mutant mice with defects in growth hormoneproduction. Proc. Natl Acad. Sci. USA 98, 6736–6741 (2001). 94. Conti, B. etal . Transgenic mice with a reduced core body temperature have an increased lifespan. Science 314, 825–828(2006). 95. Jankord, R. Herman, J. P. Limbic regulation of hypothalamo-pituitary-adrenocortical functionduring acute and chronic stress. Ann. NY Acad. Sci. 1148, 64–73 (2008). 96. Conrad, C. D.Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerabilityhypothesis. Rev. Neurosci. 19,395–411 (2008). 97. Bao, A. M., Meynen,G. Swaab, D. F. The stress system in depression and neurodegeneration: focuson the human hypothalamus. Brain Res. Rev. 57, 531–553 (2008). 98. Zahn, J. M. etal . Transcriptional profiling of aging in human muscle reveals a common agingsignature. PLoS Genet. 2,e115 (2006). 99. Rodwell, G. E. et al . A transcriptional profile of aging in the human kidney. PLoS Biol. 2, e427 (2004). 100. Maalouf, M., Rho, J. M. Mattson, M. P. The neuroprotectiveproperties of calorie restriction, the ketogenic diet, and ketone bodies. BrainRes. Rev. 59, 293–315 (2009). 志谢 因篇幅所限,许多研究和参考文献未能注明出处,谨致歉意。本项工作支持单位:美国国立卫生研究院 ( NIH ) 国立衰老研究院 ( NIA ) 、埃里森 ( Ellison ) 医学基金会、格伦 ( Glenn ) 医学研究基金会。 作者信息 复印和许可信息参见 http : //www.nature.com/reprints 。作者声明不存在经济利益冲突。通信请与作者联系 ( Email : bruce_yankner@hms.harvard.edu ) 脑老化机制与认知衰退(Neural mechanisms of ageing and cognitive decline).pdf
Role of autophagy in prion protein-induced neurodegenerative diseases Hao Yao, Deming Zhao, Sher Hayat Khan and Lifeng Yang Acta Biochim Biophys Sin (Shanghai). 2013 Jun;45(6):494-502. doi: 10.1093/abbs/gmt022 State Key Laboratories for Agrobiotechnology, Key Lab of Animal Epidemiology and Zoonosis, Ministry of Agriculture, National Animal Transmissible Spongiform Encephalopathy Laboratory, College of Veterinary Medicine, China Agricultural University, Beijing 100193, China Prion diseases, characterized by spongiform degeneration and the accumulation of misfolded and aggregated PrPSc in the central nervous system, are one of fatal neurodegenerative and infectious disorders of humans and animals. In earlier studies, autophagy vacuoles in neurons were frequently observed in neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases as well as prion diseases. Autophagy is a highly conserved homeostatic process by which several cytoplasmic components (proteins or organelles) are sequestered in a double-membrane-bound vesicle termed 'autophagosome' and degraded upon their fusion with lysosome. The pathway of intercellular self-digestion at basal physiological levels is indispensable for maintaining the healthy status of tissues and organs. In case of prion infection, increasing evidence indicates that autophagy has a crucial ability of eliminating pathological PrPSc accumulated within neurons. In contrast, autophagy dysfunction in affected neurons may contribute to the formation of spongiform changes. In this review, we summarized recent findings about the effect of mammalian autophagy in neurodegenerative disorders, particularly in prion diseases. We also summarized the therapeutic potential of some small molecules (such as lithium, rapamycin, Sirtuin 1 and resveratrol) targets to mitigate such diseases on brain function. Furthermore, we discussed the controversial role of autophagy, whether it mediates neuronal toxicity or serves a protective function in neurodegenerative disorders. 图例: 朊病毒疾病中的自噬作用 全文: http://abbs.oxfordjournals.org/content/45/6/494.full
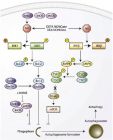
【文献导读】 不久前,Cell旗下的Molecular Cell杂志发表了英法科学家有关一氧化氮(NO)与细胞自噬、神经退行性病变的研究结果,首次报道NO可通过激酶的S-硝基化阻断细胞自噬活性,并发现NO合成的抑制能减少亨廷顿蛋白(Huntingtin)的聚集。亨廷顿蛋白的错误折叠与聚集体形成是亨廷顿病(HD)的病因,类似现象出现于帕金森病(PD)的α -突触核蛋白(α -synuclein)、阿尔茨海默病(AD)的Tau蛋白中,它们参与了神经元的变性死亡过程。这 项研究成果的意义在于初步阐明了NO与神经退行性病变的关系,为今后以NO合酶(NOS)为靶点探讨预防和治疗HD乃至PD、AD指明了方向。 作者在文章中提到细胞内NO负控制自噬的三条途径,其中一条途径是nNOS、eNOS、iNOS直接阻遏自噬(未证实),另两条途径涉及mTOR和另一条信号通路,过程较复杂,算是NO间接抑制自噬。以mTOR信号转导为例,NO先使信号通路上游的IKKβ激酶发生S-硝基化,从而减少了磷酸化IKKβ激酶数量及下游AMPK激酶的磷酸化,导致TSC2活性降低,进而缓解TSC1/2对Rheb的抑制作用,于是mTOR被激活,其结果是自噬功能受损。 下面这个示意图清楚地显示了NO阻断细胞自噬的三条途径: 虽然S-硝基化修饰对蛋白质功能具有调节作用,但它是一种可逆反应,在有巯基化合物或抗氧化剂存在时会从蛋白质上脱落下来。作者并未指出NO通过S-硝基化调节自噬是一种通用机制还是偶然现象。此前曾有两个研究小组分别发表了对于线粒体蛋白DRP的S-硝基化作用的结论完全相反的研究论文(Cho et al.Science,2009,324:102;Bossy et al.J Alzheimers Dis,2010,20:S513-S526),这是否意味着他们的实验条件有所不同,有个小组检测到S-硝基化,而另一个小组没有检测到S-硝基化? 事实上,除了S-硝基化外,蛋白质上的Tyr、Trp等也能发生硝化反应,但前提是NO与超氧阴离子(O2-)结合成超氧亚硝酸阴离子(ONOO-)。也就是说,在上面的信号通路中,每种蛋白质都有可能被硝化而影响其活性。虽然作者在文章中并未提到这种可能性,但留下了一句意味深长的话:Another possibility may be that NOS directly affacts the autophagy machinery downstream of these signaling pathways。 在老龄动物中,eNOS仍然正常表达,但NO明显不足,表明eNOS活性下降。这是什么原因呢?这主要是因为衰老促进O2-大量产生,NO与O2-反应形成ONOO-而被消耗。另外,在大量ROS作用下,eNOS可发生“解偶联”(uncoupling),即不再合成NO,而是产生O2-。因此,在讨论NO的生理功能时,要注意区分“生物可利用性NO”(bioavailable NO)和“生物不可利用性NO”(bioinavailable NO)。 从上面的理解出发,可知NO是一把“双刀刃”,好的一面是可以补充因衰老导致的NO不足,缓解血管功能失调,如“伟哥”对毛细血管的作用;不好的一面是与O2-形成有害的ONOO-,引起基因突变及其他多种生物大分子损伤。热量限制(CR)确实能通过下调O2-并上调NO延缓衰老,但假如单纯补充NO,能否促进长寿呢?大家应该能够回答这个问题。 这篇文章是免费的,任何人都可以从Cell网站下载。 原文链接:S arkar S.et al.(2011) Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 43: 19-3 2
 标签: 自噬
标签: 自噬