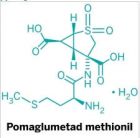
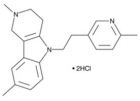
二期临床是2009年五月发表(人数199人,剂量是两组:10mg, 20mg),三期临床的文章是6月发表的,都是四军大西京医院的赵医生(G. Zhao)主持。标题上说的是多中心的研究。但是总人数不到400,而且在接受治疗的病人中,中度和重度病人占比较低,因此样本量不是很大,对于脑中风这种病种,这样的病人数好像是少了点。从临床研究数据来看,GSRD还是有效果的。该研究没有比较性研究。总的来说,数据还是单薄了点。 该研究注明的资助单位是:Tai-He Biopharmaceutical Co. Ltd. (Guangzhou, P.R. China). 另:查了一些人参皂苷的资料,三七里面人参皂苷_Rd的含量很低,单纯从植物里面提取,成本不小的。现在还没有方法进行化学合成,据现有资料表明,现在比较好的方法是首先获得各种人参皂苷的亚型,比如Rb1,Rb2等,它们的侧链和Rd不一样,需要通过一些微生物表达的酶通过酶促反应,将其转化成Rd。化学药物里面,有些药物因为合成困难,难以产业化,人参皂苷-Rd不知道这方面具体如何,需要进一步研究。GSRD为何这么受关注,是因为它是特异的受体拮抗剂,可以阻断经受体型通道进入的钙电流(查文献应该是TRP受体),这种电流一般持续时间长,对细胞损伤很大。 三期临床数据 数据来源文献: X. Liu,et al, Ginsenoside-Rd improves outcome of acute ischaemic stroke – a randomized, double-blind, placebo-controlled, multicenter trial,European Journal of Neurology, 2012,19(6):855-863. 临床实验效果统计分析采用了两种方法,一是改良型Rankin指数(Modified Rankin Scale, mRs)和NIHSS。 mRs的统计结果如下: a为全样本分析,b为有效样本分析。 mRs系统采用分级来说明病人的临床症状。级别越小,说明病人的病情症状越轻,0是没有症状,1-2基本上可以独立照顾自己,3-4为中度,生活不能完全自理,需要协助,5是严重缺乏自理能力,需要全天候陪护。 按照全样本分析数据,与对照组(Placebo)相比,人参皂苷-Rd组(GSRD)在分级上出现了一定程度的改善。0-2级比例,安慰组为64%,干预组为80%,有一定程度提升;3-4级比例,安慰组33%,干预组14%,显著下降;5-6级,安慰组5%,干预组2%,显著下降。有效样本的分布比例类似。 如果从病人症状的迁徙率上看,干预组病人症状从重症向中度,或者从中度向轻度迁徙的比率是非常明显的。该试验共有病人386人,其中对照组96人,干预组290人。该试验中由于中度和中度患者的人数较小,因此还不能很好的表明GSRD的临床效果。另外没有现有临床用药的对比,因此不能评价GSRD的相对效果是否有提升,不能说明Me Better. 其它分析方法: NIHSS(NIH中风评分,小于10为轻度)的评分,处理组有77%的患者在10分一下,安慰组为78%。腔隙性梗死(Lacunar infarct)没有明显差别。下图不大看得懂,作者说NIHSS分数在处理组有明显改善,P值小于0.01.置信区间0.95。 其它图表: 原文链接: http://onlinelibrary.wiley.com/doi/10.1111/j.1468-1331.2011.03634.x/full 临床指标简介: The Modified Rankin Scale (mRS) The scale runs from 0-6, running from perfect health without symptoms to death . 0 - No symptoms. 1 - No significant disability. Able to carry out all usual activities, despite some symptoms. 2 - Slight disability. Able to look after own affairs without assistance, but unable to carry out all previous activities. 3 - Moderate disability. Requires some help, but able to walk unassisted. 4 - Moderately severe disability. Unable to attend to own bodily needs without assistance, and unable to walk unassisted. 5 - Severe disability. Requires constant nursing care and attention, bedridden, incontinent. 6 – Dead OCSP分类法 The Oxford Community Stroke Project classification (OCSP, also known as the Bamford or Oxford classification) relies primarily on the initial symptoms. Based on the extent of the symptoms, the stroke episode is classified as total anterior circulation infarct (TACI), partial anterior circulation infarct (PACI), lacunar infarct (LACI) or posterior circulation infarct (POCI). These four entities predict the extent of the stroke, the area of the brain affected, the underlying cause, and the prognosis.
出版空间以及编辑关注度的竞争异常激烈。将原稿投送给杂志编辑,附上一封信“原稿请见附件”是远远不够的。投稿信是你与拟投杂志直接交流的机会。除了写明你的研究与众不同外,还应直接向总编辑说明为什么你的发现很重要及其应该在此杂志上发表的理由。 投 稿信应含有几个重要内容。具体内容可通过www.liwenbianji.cn/coverletter 链接下载。大家可根据批注中的建议起草你自己的投稿信,选择提出的句子类型替代括号中的句子。投稿信的格式几乎适用于所有投稿;当然,某些类别的论文需要 加入额外的内容。例如,关于临床试验数据的存储信息通常需要附上一份临床试验报告,提供你的序列数据进入公共数据库的信息。 查阅目标杂志 的《稿约》是每篇稿件的既定程序,其中很可能含有投稿信必须写入的内容。另外一个信息来源是杂志的投稿网页。尽管以下列出的内容以及关于“Edanz投稿 信模版”中描述的内容不一定完全都是这些目标杂志所要求的,但所有这些都是投稿信中必不可少的,因为这样做可引起编辑对你的关注。以下方法适用于投稿信的 撰写: • 一些杂志根据其刊出文章领域的不同进行编辑分工,你可以根据不同的领域,有时也可根据编辑的专业背景选择最合适的编辑。直接称呼收信编辑,如:“Dear Dr. Smith”。如果不能找到合适的编辑,可将投稿信写给总编辑。 • 信的开头应写出文章题目,希望文章在杂志的哪一个栏目或作为哪一个文章类别发表,以及投稿杂志的名称。 • 之后简单叙述研究背景与理论基础,说明研究目的以及开展的工作。然后简单描述研究成果。 • 接下来的段落很重要。你需要向研究界解释你的发现的意义,特别是对杂志读者的意义。如果你不能解释为什么该杂志读者会对你的发现感兴趣,你需要选择另一家 更合适的刊物,因为编辑只将他们认为会引起读者兴趣的文章送同行评议。研究一下你准备投稿杂志的“目标与刊出范围”会对你有帮助。 • 投稿信的最后一段应包含杂志所要求的声明或说明。这些通常包括关于利益冲突、基金资助与资助来源的声明,以及所有作者已阅读过并同意文章的内容以及未一稿多投的声明。每个作者的作者资格确认也是需要的。 • 最后,留下详细的通讯方式以及礼貌的结束语。 示例: 英文原文 The cover letter: your sales pitch Competition for publication space and for editors’ attention is now very high, and it is no longer sufficient to send a manuscript to a journal editor along with a letter saying little more than “please find my manuscript attached”. The cover letter is your opportunity to directly address the editor of your target journal. It can be used to set your study apart from others and directly explain to the editor why your findings are important and why they should be published in their journal. There are a number of important components of a cover letter, all of which should be included. These components are described in detail in Edanz Cover Letter Template, which is shown on the following page and can be downloaded from: www.liwenbianji.cn/coverletter. This template can be used to develop your own cover letters by following the suggestions in the comments and replacing the bracketed sentences with the types of sentences explained. The format of this letter is applicable for most if not all submissions, although additional sections may be required for some types of paper; for example, information about deposition of clinical trial data would most likely need to accompany a report of a clinical trial, and information about the deposition of sequence data into public databases would possibly need to be provided where such data has been obtained. As always, the target journal’s instructions to authors should be consulted; these will most likely outline the information that absolutely must be included in the cover letter. Another source of this information is the journal’s submission webpages. Although not all of the components listed below and described in the cover letter template will be described as required on the target journal’s webpages, all should be included in your letter, because to do so will increase your chances of grabbing the editor’s attention. The following principles apply to cover letter development: • Some journals have different editors for the different areas of research the journal covers and you can choose the most appropriate one based on area and occasionally also editor profiles. Address your letter personally to the appropriate editor, e.g., “Dear Dr. Smith”. If one cannot be readily identified, address your letter to the editor-in-chief. • Begin by providing the title of your manuscript, the section/publication type you would like to see it published as, and the name of the journal you are submitting it to. • You then need to provide a very brief background and rationale for your study, explaining why you did what you did. This can be followed by a brief description of the results. • The following paragraph is very important. You will need to explain the significance of your findings to the research community, and specifically to the readers of your target journal. If you find it difficult to explain why the readers of that journal would be interested in your findings, then you may need to select a more appropriate journal. Editors will only send papers to review that they think will be of interest to their readers. Studying the ‘aims and scope’ of your chosen journal might help with this. • The last paragraph of the letter should contain any statements or declarations required by the target journal. These usually include declarations of any conflicts of interest, grant support or other sources of funding, a statement that all authors have read and approved the manuscript and a statement that the same manuscript has not been submitted elsewhere. Confirmation of each author’s qualification for authorship may also be required. • Finally, include details for correspondence and a polite farewell. Example: Dr Daniel McGowan 分子神经学博士 理文编辑学术总监
Hi, Off label and open label are different. Off label : Getting the drug outside the trial, if you have been on it for a diffeent indication and responded , the drug is made available to you. Open label : Trial withone or two drugs where the content of the drug is known both to the patient and the doctor. No blind drug but in a trial. Drug may be available commercially on prescription as well. Dr Sharat C Misra MD,DM,FACG Re:What Here we go again: Open label , open trial : all know the drug Single Blind trial : Patient doesnt know the drug, Doc knows the drug he is giving Double blind trial : Neither the Doc nor the patient know the drug, they are labelled A and B and the codes are in a sealed envelope , decoded only after the trial, usually by the company suppying the drug or the trial co-ordinator. Dr Sharat C Misra MD,DM,FACG http://www.hbvhbv.com/forum/thread-64342-1-1.html
氢气生物学研究目前进展迅速,虽然有大量的动物实验证明对许多疾病具有治疗作用,但如果没有严格的随机对照临床试验的证据,则无法获得临床治疗特别是国家医药管理局等的最终许可,也就是说无法获得官方的正式批准用于临床的使用。开展严格对照的临床研究是氢气医学发展最重要的任务和手段。也是将来深入研究氢气生物学效应最重要的环境保障和研究目的。 但国际上在临床试验方面进展并不快,到目前为止,临床试验的报道基本都来自日本,这里是从世界卫生组织临床试验注册的信息中检索到的临床试验注册信息也说明这个问题,这些信息显示出日本在神经系统损伤治疗方面的关注度比较大,例如 6 项注册试验中有 3 项属于神经系统损伤治疗效果的研究,分别是中风、巴金森病和中度认知障碍的研究。比较有意思的是,最早报道氢气生物学效应的日本医科大学没有注册临床试验的信息,是他们没有信心,还是没有获得研究经费的资助。因为他们曾经获得来自商业公司的赞助,并成立氢气医学中心。从这一点上看,似乎没有这些问题。但内情不清楚。 第一项:氢气水治疗巴金森病 第二项:氢气水对正常人的抗氧化效果评价 第三项 氢气生理盐水注射对脑缺血的治疗效果评价 第四项 氢气水对中度认知障碍治疗效果的研究 第五项 氢气水治疗间质性膀胱炎 第六项 氢气水对糖尿病的治疗效果评价 所有信息可从世界卫生组织的临床试验注册网上免费检索,更详细的信息可从 http://apps.who.int/trialsearch/AdvSearch.aspx 检索。 建议检索词为 : hydrogen 。 Recruitment status 选择 ALL 。否则无法获得全面的信息。 详细信息 第一项:氢气水治疗巴金森病 2012 年 3 月 14 日注册的用“氢气水治疗巴金森病”开始实验 2010 年 1 月 1 日顺天堂大学附属医院神经外科,联系人 Asako Yoritaka 。日本学者曾经报道使用氢气水治疗巴金森病的动物实验效果,发表论文 3 篇。全部使用自由饮用氢气水。治疗设计 The subjects should make 1000 ml of molecular hydrogen water which contains 1.6 ppm dissolved hydrogen by Aquerable, and consume for 48 weeks . Placebo water which is not contained molecular hydrogen water made from pseudo-machine. The subjects consume for48 weeks. 第二项:氢气水对正常人的抗氧化效果评价 Studies on in vivo effects of drinking a water product dissolving hydrogen gas as an in vitro antioxidant additive 杏林大学 Atsushi Hiraoka 自 2009 年 5 月 1 日开始的针对健康人的一项研究,排除肝脏肾脏功能异常和月经期女性。 Ingestion of 500ml per day of hydrogen gas-dissolving water for 1 week.Ingestion of 500ml per day of control water without hydrogen gas for 1 week.. 观察指标 the levels of serum LPO and urine 8-OHdG in subjects immediately before and after 1-week drinking period for 500ml per day of tap water with or without dissolved hydrogen gas at 0.34mg/l and 1-week before and after the drinking per day. 第三项 氢气生理盐水注射对脑缺血的治疗效果评价 日本国防医科大学神经外科 Hiroshi Nawashiro 于 2011/06/01 开始的 Molecular hydrogen for ischemic stroke 。选择诊断后症状发生 24 小时内脑缺血患者 Patients were eligible for enrollment if they were 18 years or older and had a clinical diagnosis of acute ischemic stroke within 24 hours of symptom onset. They had to score at least 6 on the National Institutes of Health Stroke Scale (NIHSS) with at least 2 points for limb weakness. All patients received appropriate routine stroke care as per local treatment practices, including alteplase for eligible patients presenting 3 hours from onset; patients receiving alteplase had to commence the study drug before the alteplase infusion.Exclusion criteria: Patients with acute ischemic stroke beyond 24 hours of symptom onset. 治疗方法为静脉点滴注射氢气生理盐水。效果评价 modified Rankin scale (mRS) (days 7, 30, and 90), the NIHSS (days 7 and 90), and the Barthel Index (days 7, 30, and 90) Safety Assessments Vital signs were recorded at enrollment and at specified times throughout the infusion and follow-up periods. Routine laboratory data and ECGs were performed at the time of enrollment, at 24 and 72 hours, and on day 7 and were analyzed centrally (ECGs at day 7 were performed only if abnormal at 72 hours). To assess any effect of hydrogen on hemorrhagic transformation after alteplase administration, brain imaging was repeated after 72 hours in patients who were receiving concomitant treatment with alteplase. Symptomatic hemorrhagic transformation was defined as an increase in the NIHSS score of at least 4 points within 36 hours, plus evidence of any blood on neuroimaging after treatment with alteplase. Patients meeting criteria for progressive stroke (NIHSS increase of 4 points within 72 hours) or new stroke in the first week were also reimaged. 第四项 氢气水对中度认知障碍治疗效果的研究 筑波医科大学临床医学研究所神经精神学系 Takashi Asada2009/07/01 开始的 A randomized trial to assess the effects of hydrogen-ride dissolution water for the patients with mild cognitive impairment (MCI). 中度认知障碍的研究。 招募对象: Inclusion criteria: 1) Participants of the Tone project. 2) Being able to give a written informed consent to the participation in the present study. 3) Having diagnosis of the mild cognitive impairment. 4) Being able to observe the following requirement: good compliance with the water; participation in the scheduled examinations for assessment; keeping a log-diary recording the consumption of the water. 5) Having a modified Hachinski Ischemic score of 4 or less. 6) Having the 15-item Geriatric Depression Scale score of 6 or less. Exclusion criteria: 1) Meeting DSM-IV TR criteria for dementing illnesses. 2) Having serious or unstable illnesses. 3) Having a history within past 5 years of serious infectious disease affecting the brain and/or malignant diseases. 4) Having a history of alcohol or drug abuse or dependence (on DSM-IV TR) within the past 5 years. 5) Receiving any types of anti-Alzheimer drugs. 6) Recent (within 4 weeks) initiation of medications that affect the central nervous system.Age minimum: 67years-old.Age maximum: Not applicable Gender: Male and Female 研究内容: mild cognitive impairment 治疗手段 The patients of hydrogen group will be intervened with 500ml hydrogen dissolution water every-day for 2 years. The patients of placebo group will be intervened with 500ml ordinary water every-day for 2 years. 效果评价 Score in Japanese version of ADAS-Cog and Mini Mental State Examination. Scores in Japanese version of ADCS-ADL, MRI and SPCET imaging, and Geriatric Depression Scale. 第五项 氢气水治疗间质性膀胱炎 Koushinkai Hospital 的 Comprehensive Support Project for Clinical Research Office 于 2008/07/01 开始的 A randomized trial to asses the effects of hydrogen-rich dissolution water in patients with interstitial cystitis 。至少现在没有氢气在间质性膀胱炎方面的研究报道,无论是动物实验还是临床报道。 研究对象标准: Inclusion criteria: 1) Patients who are able to give written informed consent 2) Patients who has the characteristic finding under hydraulic distension of the bladder in interstitial cystitis by cystoscope 3) It has taken more than 12 weeks after patients took the hydraulic distension of the bladder, and symptom of patients are in stable. 4) More than 7 marks in total of Interstitial Cystitis Score in registration 5) More than 4 marks in Q4 (degree of bladder pain) in Interstitial Symptom Score 6) Age is over 20 years and less than 80 years 7) Patients are able to do the following things in this trial; getting good compliance with intaking investigating food and coming to hospital, and writing the diary and the questionnaire accurately by themselvesExclusion criteria: 1) More than 200ml of an average voided volume at a time before the registration 2) Patients with active infection of urinary tract 3) Patients with bacterial cystitis within 12 weeks before registration 4) Patients with vaginosis 5) Patients with calculus of lower urinary tract or urethral diverticulum 6) Patients with nephrosis syndrome 7) Patients with active genital herpes 8) Patients who have operated the surgery in pelvis or its circumference and it has not taken more than 24 hours after the surgery 9) Patients with cerebrospinal disease 10) Patients with the follow disease or suspected disease; neurogenic bladder, cystitis radiation, tuberculous cystitis, cystitis with BCG, drug associated cystitis 11) Start, stop, or change of the dose of the following drugs within 4 weeks after the registration; (a) Antiphlogistic analgetic (b) Antidepressant (c) Anticholinergic drug (d) Antihistamine drug (e) Ataractic drug (f) Drug treatment for frequent urination and acraturesis (g) Steroid drug 12) Start or stop new bladder training or diet therapy within 4 weeks befor registration 13) Patients who has received bladder instillation therapy, electrical stimulation therapy, or acupuncture and moxibustion within 12 weeks before registration 14) Patients with serious hepatic or kidney damage 15) Patients with serious heart disease 16) Patients with malignant tumors which effect their general status or survival time 17) Patients with the history of serious drug-induced adverse effect 18) Patients who are in pregnancy, while breast-feeding, or have possibilities of them, or desire pregnancy in test period 19) Patients who have taken part in the her clinical research within 12 weeks 20) Patients who have taken part in the her clinical research within 12 weeks 21) Patients who are inadequate, which their physicians assessed itAge minimum: 20years-old Age maximum: 80years-oldGender: Male and Female 治疗方法 The patients will be intervened with hydrogen dissolution water group (hydrogen group) 200ml every three times in a day in 2 months (56days) . After that, the patients in hydrogen dissolution water group will be transferred to the additional intervention term after the end of intervention.And after that, the patients will be randomized to withdrawal terms for more 1 month with hydrogen dissolution water or with placebo water. The proportion of the patients who has been assessed "success" at the end of the intervention Secondary Outcome(s) 1) Changes of the Symptom Score in Interstitial Cystitis Symptom Index(ICSI) 2) Changes of the Problem Score in Interstitial Cystitis Problem Index(ICPI) 3) An Average frequency of urination per day 4) An Average voided volume at a time 5) Degree of urge to urinate; PUF symptom score 6) Degree of bladder pain 7) Impression by patients with GRA (Global Response Assessment) 8) Urine Test; 8-OHdG in urine 9) Adverse Events (we cannot deny the association between the food and the event) 第六项 氢气水对糖尿病的治疗效果评价 研究京都大学医学院 Comprehensive Support Project for Clinical Research Office 于 21/08/2008 开始的 A Randomized trial to assess the effects of hydrogen-rich dissolution water for patients with impaired glucose tolerance or impaired fasting glucose 。该研究已经发表论文。 研究对象标准: Inclusion criteria: 1) Patients who are abele to give written informed consent 2) FBS is over 100mg/dl and under 126mg/dl in registration 3) Age is over 20 years and less than 80 years 4) Type of practice: outpatient department 5) Patients are able to do the following things in this trial - getting good compliance with consuming investigational food and coming to hospital, and writing the diary and the questionnaire accurately by themselvesExclusion criteria: 1) Patients who have receive drug treatment for diabetes 2) Patients with the diseases which have possibility with impaired glucose tolerance 3) Patients with serious liver or kidney damage 4) Patients with serious heart disease or cerebrovascular disorders 5) Patients with serious disease in pancreas or blood disease 6) Patients with malignant tumors which effect their general status or survival time 7) Patients who are in pregnancy, while breast-feeding, have possibilities of them, or desire pregnancy in test period 8) Patients with alcohol abuse 9) Patients who have taken part in the her clinical research within 12 weeks 10) Patients who have taken part in the her clinical research within 12 weeks 11) Patients who are inadequate, which their physicians assessed it Age minimum: 20years-oldAge maximum: 70years-oldGender: Male and Female 研究内容 Impaired glucose tolerance or impaired fasting glucose 治疗手段 The patients will be intervened with hydrogen dissolution water group (hydrogen group) 200ml every three times in a day in 3 months (84days) . The patients will be intervened with normal water group (placebo group) 200ml every three times in a day in 3 months (84days) 效果评价方法 1)75gOGTT(glucose);0 minute (in the fasting state),30,60,90 minutes later after loading2)Delta AUC(0-120min);The difference of the area under the plasma glucose concentration before and after administration
出版空间以及编辑关注度的竞争异常激烈。将原稿投送给杂志编辑,附上一封信“原稿请见附件”是远远不够的。投稿信是你与拟投杂志直接交流的机会。除了写明你的研究与众不同外,还应直接向总编辑说明为什么你的发现很重要及其应该在此杂志上发表的理由。 投 稿信应含有几个重要内容。具体内容可通过www.liwenbianji.cn/coverletter 链接下载。大家可根据批注中的建议起草你自己的投稿信,选择提出的句子类型替代括号中的句子。投稿信的格式几乎适用于所有投稿;当然,某些类别的论文需要 加入额外的内容。例如,关于临床试验数据的存储信息通常需要附上一份临床试验报告,提供你的序列数据进入公共数据库的信息。 查阅目标杂志 的《稿约》是每篇稿件的既定程序,其中很可能含有投稿信必须写入的内容。另外一个信息来源是杂志的投稿网页。尽管以下列出的内容以及关于“Edanz投稿 信模版”中描述的内容不一定完全都是这些目标杂志所要求的,但所有这些都是投稿信中必不可少的,因为这样做可引起编辑对你的关注。以下方法适用于投稿信的 撰写: • 一些杂志根据其刊出文章领域的不同进行编辑分工,你可以根据不同的领域,有时也可根据编辑的专业背景选择最合适的编辑。直接称呼收信编辑,如:“Dear Dr. Smith”。如果不能找到合适的编辑,可将投稿信写给总编辑。 • 信的开头应写出文章题目,希望文章在杂志的哪一个栏目或作为哪一个文章类别发表,以及投稿杂志的名称。 • 之后简单叙述研究背景与理论基础,说明研究目的以及开展的工作。然后简单描述研究成果。 • 接下来的段落很重要。你需要向研究界解释你的发现的意义,特别是对杂志读者的意义。如果你不能解释为什么该杂志读者会对你的发现感兴趣,你需要选择另一家 更合适的刊物,因为编辑只将他们认为会引起读者兴趣的文章送同行评议。研究一下你准备投稿杂志的“目标与刊出范围”会对你有帮助。 • 投稿信的最后一段应包含杂志所要求的声明或说明。这些通常包括关于利益冲突、基金资助与资助来源的声明,以及所有作者已阅读过并同意文章的内容以及未一稿多投的声明。每个作者的作者资格确认也是需要的。 • 最后,留下详细的通讯方式以及礼貌的结束语。 示例: 英文原文 The cover letter: your sales pitch Competition for publication space and for editors’ attention is now very high, and it is no longer sufficient to send a manuscript to a journal editor along with a letter saying little more than “please find my manuscript attached”. The cover letter is your opportunity to directly address the editor of your target journal. It can be used to set your study apart from others and directly explain to the editor why your findings are important and why they should be published in their journal. There are a number of important components of a cover letter, all of which should be included. These components are described in detail in Edanz Cover Letter Template, which is shown on the following page and can be downloaded from: www.liwenbianji.cn/coverletter. This template can be used to develop your own cover letters by following the suggestions in the comments and replacing the bracketed sentences with the types of sentences explained. The format of this letter is applicable for most if not all submissions, although additional sections may be required for some types of paper; for example, information about deposition of clinical trial data would most likely need to accompany a report of a clinical trial, and information about the deposition of sequence data into public databases would possibly need to be provided where such data has been obtained. As always, the target journal’s instructions to authors should be consulted; these will most likely outline the information that absolutely must be included in the cover letter. Another source of this information is the journal’s submission webpages. Although not all of the components listed below and described in the cover letter template will be described as required on the target journal’s webpages, all should be included in your letter, because to do so will increase your chances of grabbing the editor’s attention. The following principles apply to cover letter development: • Some journals have different editors for the different areas of research the journal covers and you can choose the most appropriate one based on area and occasionally also editor profiles. Address your letter personally to the appropriate editor, e.g., “Dear Dr. Smith”. If one cannot be readily identified, address your letter to the editor-in-chief. • Begin by providing the title of your manuscript, the section/publication type you would like to see it published as, and the name of the journal you are submitting it to. • You then need to provide a very brief background and rationale for your study, explaining why you did what you did. This can be followed by a brief description of the results. • The following paragraph is very important. You will need to explain the significance of your findings to the research community, and specifically to the readers of your target journal. If you find it difficult to explain why the readers of that journal would be interested in your findings, then you may need to select a more appropriate journal. Editors will only send papers to review that they think will be of interest to their readers. Studying the ‘aims and scope’ of your chosen journal might help with this. • The last paragraph of the letter should contain any statements or declarations required by the target journal. These usually include declarations of any conflicts of interest, grant support or other sources of funding, a statement that all authors have read and approved the manuscript and a statement that the same manuscript has not been submitted elsewhere. Confirmation of each author’s qualification for authorship may also be required. • Finally, include details for correspondence and a polite farewell. Example: Dr Daniel McGowan 分子神经学博士 理文编辑学术总监
分享一下2012年5月16日Biotronik公司在其网站上发布他们对镁合金药物洗脱支架的12个月临床(欧洲多中心,6个月和12个月2组共46个患者)随访结果,与去年发布的6个月结果相比,Twelve-month results from the BIOSOLVE-I study with the DRug Eluting Absorbable Metal Scaffold (DREAMS) demonstrate safety and confirm vessel vasomotion.这对做可降解金属的同仁们会是个鼓励。 pdf下载在这里 BIOTRONIK_PR%20BIOSOLVE-I_EuroPCR_EN.pdf http://www.biotronik.de/wps/wcm/connect/int_web/biotronik/newsroom/press_releases?p=http://www.biotronik.de/wps/wcm/connect/int_web/biotronik/newsroom/press_releases/press_release_biosolve_ipw=770pt = At one year, DREAMS demonstrated a low 7.0% rate of target lesion failure with no death or scaffold thrombosis. The target lesion revascularization rate was 4.7%. The analysis showed an in-scaffold late lumen loss of 0.52 mm, which was further reduced from the 6-month results. Vasoreactivity at 12 months did not significantly change from the 6-month results, suggesting that the natural physiology of the vessels was already restored after 6 months of healing and could be sustained. “The reduction of the 12-month late lumen loss is likely due to plaque regression and late expansive remodeling. At 6 months, we have already seen that vasomotion is restored, so the new results verify that the vessels are naturally adapting to accommodate flow,” said Professor Haude. “Additionally, we could further validate that the return of natural vessel angulation that we saw at 6 months was maintained also at 12 months. These results suggest a real restoration of the vessel’s natural architecture.” DREAMS is made of a proprietary magnesium alloy coated with a matrix of a bioabsorbable polymer and paclitaxel to inhibit neointimal cell proliferation within the first few months after scaffold implantation. Mechanical properties of the magnesium alloy allow deployment of the device to be comparable to conventional metallic stents. “The particularly positive feature of the magnesium scaffold is its nice conformability to the vessel wall. A few months after DREAMS implantation, the artery is able to regain its original shape and physiology. This is the main benefit of the magnesium scaffold over the more rigid polymer-based platforms,” Professor Haude added. “These positive one-year results confirm that we are working in the right direction. BIOTRONIK continues to invest in our DREAMS program as we believe that bringing vascular restoration therapy to patients without changing the way physicians implant a scaffold is important to the adoption of this technology,” stated Alain Aimonetti, Vice President of Sales and Marketing at BIOTRONIK Vascular Intervention. “An improvement of late lumen loss between 6 and 12 months shows us that the values become more predictable over time. It makes sense, especially for the bioabsorbable scaffolds, to focus on long-term outcomes and not so much on short-term results,” Aimonetti concluded. “We see huge potential for DREAMS since it combines deployment and postdilatation properties and long-term outcomes comparable to DES with the additional benefits of vascular restoration therapy. About the BIOSOLVE-I Clinical Trial The BIOSOLVE-I first-in-man study is a prospective, multicenter, nonrandomized, European, first-in-man clinical trial evaluating the safety and efficacy of DREAMS. Forty-six patients were enrolled in two cohorts that evaluated the primary endpoint of target lesion failure (TLF) at 6 months for cohort 1 and at 12 months for cohort 2. About the DRug Eluting Absorbable Metal Scaffold (DREAMS) DREAMS is part of a revolutionary new treatment option for patients with coronary artery disease . In contrast to existing permanent stents, this device is based on a slow-absorbing, high-strength magnesium backbone. The vessel is not caged as it would be with a permanent implant, thus the artery is able to resume its natural physiology. This novel treatment concept opens a new area of vascular restoration therapy. Properties of the proprietary magnesium alloy allow a straightforward deployment of the device, comparable to conventional metallic stents. BIOTRONIK’s DREAMS is coated with antiproliferative paclitaxel to inhibit excess tissue growth during the healing process. The company has announced next-generation platforms under development using the BIOlute coating from the Orsiro hybrid drug-eluting stent featuring sirolimus as the active compound. About BIOTRONIK SE Co. KG As one of the world’s leading cardiovascular medical device companies, with several million implanted devices, BIOTRONIK is represented in over 100 countries with its global workforce of more than 5,600 employees. Known for having its fingers on the pulse of the medical community, BIOTRONIK assesses the challenges physicians face and provides the best solutions for all phases of patient care, ranging from diagnosis and treatment to patient management. Quality, innovation and clinical excellence define BIOTRONIK and its growing success—and deliver confidence and peace of mind to physicians and their patients worldwide.
 标签: 临床试验
标签: 临床试验