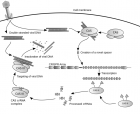
最近,随着基因编辑技术不断曝光,这项技术也越来越火,越来越受到关注,但大部分媒体都只是浅尝辄止进行宣传而已,实质性的怎么操作,具体的步骤却很少涉及。鉴于此,师兄就把张峰的手稿扒拉出来,分享给大家。同时也欢迎大家与我探讨,我的微信号:shixiongcoming,向我申请的时候注意,写上你的理由哦,最近加我的人太多,不注明来由的读者,别怪师兄拒绝你呦。 Genome Engineering Using CRISPR-Cas9 System Le Cong and Feng Zhang Abstract The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 system is an adaptive immune system that exists in a variety of microbes. It could be engineered to function in eukaryotic cells as a fast, low-cost, effi cient, and scalable tool for manipulating genomic sequences. In this chapter, detailed protocols are described for harnessing the CRISPR-Cas9 system from Streptococcus pyogenes to enable RNA-guided genome engineering applications in mammalian cells. We present all relevant methods including the initial site selection, molecular cloning, delivery of guide RNAs (gRNAs) and Cas9 into mammalian cells, verification of target cleavage, and assays for detecting genomic modification including indels and homologous recombination. These tools provide researchers with new instruments that accelerateboth forward and reverse genetics efforts. Key words Genome engineering , Cas9 , CRISPR , CRISPR-Cas9 , PAM , Guide RNA , sgRNA , DNA cleavage , Mutagenesis , Homologous recombination , Streptococcus pyogenes 1 Introduction 1.1 General Principle of Genome Engineering with Designer Nucleases Genome engineering is a highly desirable technology in biologicalresearch and biomedical applications, allowing us to change genomic sequences at will to control the fundamental geneticinformation within a cell. It has been implemented in creatingmodel cell lines or organisms for the study of gene functions orgenetics of human diseases, and in gene therapy to correctdeleterious genomic changes or confer beneficial ones. Genome engineering using engineered nucleases has been one of the mostdesignable and scalable paths to achieve precise editing of genomicsequences. The basic principle for this process could be summarizedin to a two-step process (Fig. 1 ). The first step is the introductionof targeted DNA cleavage, in the form of typically double strandbreaks (DSBs) or possibly single strand nicks (SSNs), through theactivity of nucleases or nickases, respectively. The second stepinvolves DNA repair process activated by the targeted cleavage,carried out normally by the endogenous DNA damage repair machinery within the cells. In this step, two major pathways couldbe employed, achieving different types of genome modifi cation. One pathway is to repair the cleaved site via nonhomologous endjoining (NHEJ) pathway, which applies only to the DSBs as SSNsare thought to be unable to induce NHEJ pathway. During NHEJmediatedrepair, insertion/deletion (indels) will be engineeredinto the target site. The other pathway requires the artifi cial supplyof a repair template with chosen sequence alterations into the cellin addition to the DSBs or SSNs, so that homology-directed repair(HDR) pathway would be activated. The latter pathway requireshomologous recombination (HR) between chromosomal DNAand the supplied foreign DNA stimulated by the local DNAcleavage, resulting in virtually any type of desired editing throughreplacement of native sequence by the designed HR template . Overall, one of the most essential rate-limiting steps in thistype of genome engineering technology is the targeted cleavage ofendogenous genome. Hence, effort that centers on the developmentof better designer DNA-cleaving enzymes has been a focus ofthis fi eld. In the past decades, genome editing tools based onsequence-specifi c nucleases and DNA-binding proteins such aszinc-fi nger nucleases (ZFNs) , meganucleases , and transcriptionactivator like effectors (TALEs) , among others,have opened up the possibility of performing precise perturbationof genome at single-nucleotide resolution. Recently, the emergenceof a new type of genome engineering technology based onthe Clustered Regularly Interspaced Short Palindromic Repeats(CRISPR) system, a microbial adaptive immune system, has transformedthe fi eld because of its easiness to design, rapidity to implement,low-cost, and scalability in eukaryotic cells. The CRISPR locus in microbes typically consists of a set ofnoncoding RNA elements and enzymes that harbors the ability torecognize and cleave foreign nucleic acids based on their sequencesignature. Three known CRISPR systems, types I, II, and III, havebeen described so far . Among these different CRISPRsystems, type II CRISPR systems usually only require a single protein,called Cas9, to perform the target cleavage. Recent years, thetype II CRISPR-Cas9 systems have been studied by several groupsin their native microbial domain to elucidate their molecular organizationsand functions . These studies demonstrated that Cas9 is a RNA-guided nuclease that is capable of binding to a targetDNA and introduce a double strand break in a sequencespecific manner, and its specifi city is determined by sequenceencoded within the RNA components. Following these breakthroughs, this new family of RNA guide dnucleases has been successfully developed into a multiplex genome engineering system that is simple to design, efficient, andcost-effective for mammalian genome editing purposes . Application of this system could enable introduction of different type of modifi cations, such as mutation, insertion, deletion, oreven larger chromosomal changes, through NHEJ or HDR pathways,in many different cell types and organisms . Fig. 1 Principle for genome engineering using designer nucleases or nickases. DNA cleavage induced bydesigner nucleases or DNA nicks created by designer nickases can result in double strand break (DSB), singlestrand nick (SSN), or double nicks (DNs) ( top ). These site-specifi c DNA cleavages can be repaired via two differentpathways ( bottom ). In the error-prone NHEJ pathway for DSB or DNs, the ends of the breaks are processedby endogenous DNA repair machinery and rejoined, which can result in random indels at the site ofjunction. The other pathway is the HDR pathway for both DSB, SSN, and DNs, where a repair template is supplied,allowing precise editing of the endogenous genome sequences. Parts of this fi gure are adapted from 1.2 Genome Engineeringwith CRISPR- Cas9 System The required components for CRISPR-Cas9 mediated DNA cleavageare the Cas9 protein and its bound RNA component thatguide the Cas9 to target sequence. The RNA component consistsof a RNA duplex formed by partial pairing between the CRISPRRNA (crRNA), which includes the guide sequence that bound totarget DNA through Watson–Crick base-pairing to specify the siteof cleavage, and the trans-activating crRNA (tracrRNA), whichfacilitate the maturation of crRNA and the loading of crRNA ontoCas9 (Fig. 2a ) . Our previous work demonstrated that by harnessing the well-understood CRISPR-Cas9 system from Streptococcus pyogenes SF370, we could develop a system that is sufficient for carrying out site-specifi c DNA cleavage in mammaliancells . A simplifi ed version of this design is achieved by fusionof the mature form of crRNA and tracrRNA into a chimeric singleguide RNA (sgRNA), which demonstrated even superior effi ciencycompared with the split design . Hereafter we centeron this two-component design, which enhances the convenienceof using the CRISPR-Cas9 system and multiplexing of genome targeting applications. Fig. 2 Schematic of the type II CRISPR-mediated DNA double-strand break and thedesign of chimeric sgRNA. ( a ) The type II CRISPR locus from Streptococcus pyogenesSF370 contains a cluster of four genes, Cas9, Cas1, Cas2, and Csn1, as well as twononcoding RNA elements, tracrRNA and a characteristic array of repetitive sequences(direct repeats) interspaced by short stretches of non- repetitive sequences (spacers,30 bp each). Each spacer is typically derived from foreign genetic material (protospacer),and directs the specifi city of CRISPR- mediated nucleic acid cleavage. In thetarget nucleic acid, each protospacer is associated with a protospacer adjacentmotif (PAM) whose recognition is specifi c to individual CRISPR systems. The TypeII CRISPR system carries out targeted DNA double-strand break (DSB) in sequentialsteps. First, the pre-crRNA array and tracrRNA are transcribed from theCRISPR locus. Second, tracrRNA hybridizes to the direct repeats of pre-crRNAand associates with Cas9 as a duplex, which mediates the processing of thepre-crRNA into mature crRNAs containing individual, truncated spacer sequences.Third, the mature crRNA:tracrRNA duplex directs Cas9 to the DNA target consisting of theprotospacer and the requisite PAM via heteroduplex formation between the spacer region of the crRNA and theprotospacer DNA. Finally, Cas9 mediates cleavage of target DNA upstream of PAM to create a DSB within the protospacer. ( b ) Design of chimeric sgRNA for genome cleavage. The SpCas9 nuclease ( yellow ) binds to genomic DNAdirected by the sgRNA consisting of a 20-nt guide sequence ( blue ) and a chimeric RNA scaffold ( red ). The guidesequence base-pairs with protospacer target ( blue ), located upstream of a requisite “NGG” called protospaceradjacent motif (PAM; colored in pink ). The SpCas9 mediates a DSB around 3 bp upstream to the 5′ end of the PAM( red triangles ). Parts of this fi gure are adapted from The guide sequence within sgRNA has a length of 20 bp andis the exact complementary sequence of the target site within thegenome, sometimes referred to as “protospacer” following theconvention of microbial CRISPR research (Fig. 2a ). In choosingthe target site, it is important to note the requirement of havingthe “NGG” trinucleotide motif, called protospacer adjacent motif(PAM), right next to the protospacer target on the 3′ end. TheCas9 cleavage site is located within the protospacer and positionedat around 3 bp upstream the 5′ end of the PAM, indicating apotential anchor role of the PAM for Cas9-mediated DNA cleavage(Fig. 2a, b ). The PAM sequence depends on the Cas9 proteinemployed in the CRISPR-Cas9 system, and hence, the “NGG”PAM sequence applies specifi cally to the Streptococcus pyogenesCas9 (SpCas9) discussed in this protocol. The Cas9 enzyme typically contains two nuclease domains: theRuvC-like (N-terminal RNase H fold) domain and the HNH(McrA-like) domain, each responsible for cleavage of one strand ofthe duplex DNA molecule. Therefore, there are two different versionsof Cas9 that can be used for genome modifi cation, the wildtype Cas9 that induces DSBs to the target site, and the nickaseversion of Cas9 (Cas9n) bearing a deactivating mutation in one ofthe two nuclease domains, which nicks the target DNA on onestrand. For SpCas9 that we described in this protocol, the twoderived nickases are SpCas9n (D10A) and SpCas9n (H840A),with mutation at one of the two catalytic residues of the nucleasedomain, D10 and H840, respectively. Whereas the wild typeSpCas9 is highly effi cient in executing NHEJ- and HDR-mediatedgenome engineering, its off-target effects due to the induction ofDSBs at mismatched targets within the genome cannot beneglected . The NHEJ-incompetent SpCas9n, in particularin the form of a double-nicking design where a pair of sgRNAscooperates two adjacent nickings at target genomic locus, might besuperior in terms of editing accuracy as it maintains the ability toachieve comparable genome engineering capacity and effi ciencywhile reducing the likelihood of off-target NHEJ events . Furthermore, if the catalytic residues within the two domains are simultaneously mutated, the deactivated non-cleaving version ofSpCas9 (dSpCas9) can serve as a generic RNA-guided DNAbindingprotein, priming additional applications in mammaliancells such as targeted transcriptional modulation . Morerecently, one study showed that by shortening the guide lengthwithin the sgRNA from 20 to 17 bp, the genome targeting specificity of wild type SpCas9 protein could be improved . Shortlyafterwards, two independent pieces of work demonstrated the utilizationof Cas9-FokI fusion protein as a designer dimer nucleasefor improved Cas9 specifi city . All these work on SpCas9-based system and earlier demonstration that orthogonal Cas9 proteinsfrom different microbial species can be adapted for mammaliangenome targeting , implicate the potential of optimizing theguide RNA and the Cas9 protein as valid strategies to address theoff-target issue of CRISPR-Cas9 system, raising the possibility thatan effi cient and specifi c CRISPR-Cas9 system will be realized forpowerful and precision genome engineering. Here we describe a detailed protocol for applying the CRISPRCas9 system from Streptococcus pyogenes for genome engineeringpurposes in mammalian cells. The protocol includes steps arrangedin order of actual experiments: selection of targets and cloning oftargeting constructs (Subheading 3.1 ), delivery into mammaliancells (Subheading 3.2 ), assay for genomic cleavage (Subheading 3.3 ),implementation of homologous recombination using CRISPRCassystem (Subheading 3.4 ), and the verifi cation and quantifi cationof HR effi ciency (Subheading 3.5 ). There are still numerouschallenges related to effi ciency of homologous recombination, offtargeteffects, and multiplexed targeting before we could deployCRISPR-Cas9 system to achieve robust, effi cient, and accurategenome engineering in all type of biological systems, especially forin vivo settings. Nonetheless, the general principle and designguidelines for SpCas9 in this chapter should be relevant for thesefuture efforts as well. 2 Materials 2.1 Molecular Biology Reagents 1. Backbone plasmids: pX330 (U6-chimeric guide RNA + CBh-SpCas9 backbone, Addgene ID 42230), pX335 (U6-chimericguide RNA + CBh-SpCas9n backbone, Addgene ID 42335). These and additional backbone plasmids mentioned in theprotocol are all available from Addgene, at www.addgene.org . 2. DNA oligos (Standard de-salted) for cloning of chosen guides. For oligos longer than 60 nt, the ultramer oligo (IntegratedDNA Technology, Coralville, IA, USA) is recommended. 3. PCR reagents: Herculase II fusion polymerase (Agilent, SantaClara, CA, USA). Other high-fi delity polymerases, such asKapa HiFi (Kapa Biosystems, Wilmington, MA, USA) can alsobe used for PCR in this protocol. 4. Cloning enzymes: FastDigest Bbs I, FastDigest Age I, FastAP(Thermo Scientifi c/Fermentas, Pittsburg, PA, USA), Plasmid-Safe exonuclease (Epicentre, Madison, WI, USA), T7 DNA ligase. 5. 10× T4 DNA ligase buffer. 6. 10× Tango buffer (Thermo Scientifi c/Fermentas). 7. DTT (DL-Dithiothreitol; Cleland’s reagent). 8. 10 mM Adenosine 5′-triphosphate. 9. Ultrapure water (RNAse/DNAse-free). 10. Tris-EDTA (TE) buffer. 11. E. coli competent cells. Strains such as Stbl3 (Life Technologies,Carlsbad, CA, USA) or other recombination-defi cient strain isrecommended to avoid recombination of repetitive elementsin the plasmids. 12. Luria Broth (LB) media. 13. Ampicillin. 14. Spin miniprep kit. 15. Plasmid Midi/Maxi Kit. 16. Standard gel electrophoresis reagents and apparatus. 2.2 Cell Culture and Processing Reagents 1. Cell line: human embryonic kidney (HEK) 293FT cell line. 2. Dulbecco’s Modifi ed Eagle’s Medium (DMEM). 3. 10 % fetal bovine serum (FBS). 4. GlutaMAX solution (Life Technologies). 5. 100× Pen-Strep: 100 U/μL penicillin, 100 μg/μL streptomycin. 6. OptiMEM medium (Life Technologies). 7. TrypLE® Express Enzyme (Life Technologies). 8. Transfection reagent: Lipofectamine 2000 (Life Technologies)can be used for HEK 293FT or Neuro-2a cell lines with thisprotocol. Other transfection reagents can also be used dependingon the cell type and after trial/optimization. 9. 24-well tissue culture plates. 10. pCMV-EGFP plasmid (Addgene, Cambridge, MA, USA). 11. pUC19 plasmid (NEB). 2.3 Genome Modifi cation and Homologous Recombination Assay Reagents 1. 1× PBS. 2. QuickExtract? DNA extraction kit (Epicentre). 3. SURVEYOR Mutation Detection Kit (Transgenomic, Omaha,NE, USA). 4. 4–20 % Novex TBE polyacrylamide gels and accompanyinggel-running reagents (Life Technologies). 5. PCR primer for amplifi cation of genomic DNA used inSURVEYOR assay and HR assay. 6. PCR reagents: Herculase II fusion polymerase (Agilent). Otherhigh-fi delity polymerases, such as Kapa HiFi (Kapa Biosystems)can also be used for PCR in this protocol. 7. 10× Taq PCR buffer. 8. Novex Hi-Density TBE sample buffer (Life Technologies). 9. 10,000× SYBR Gold nucleic acid gel stain (Life Technologies). 10. PCR purifi cation kit. 11. Gel extraction kit. 12. Restriction enzyme for HR assay depends on the design of theHR template. 3 Methods Ultrapure water is used for all the steps below. The setup of the experimentsis carried out at room temperature unless otherwise stated. 3.1 Design and Cloning of Genome Engineering Constructs Below is a step-wise protocol for cloning and verifi cation of targetingvectors. Prior to the cloning of the constructs for genome engineering,the target sites should be selected as described inSubheading 4 ( see Notes 1 – 3 ). 1. Prepare sgRNA oligo duplex. Resuspend the oligos ( see Note 2 ) to a fi nal concentration of 100 μM with TE buffer. Mix regents listed below to preparethe reaction mixture for each pair of oligos to phosphorylateand anneal them into ligation-ready duplex DNA fragments. Place the mixture in a thermocycler using the followin parameters to phosphorylate and anneal the oligos. Add 2 μl of annealed oligos into 398 μl of water to dilutethe products 200-fold. 2. Cloning of targeting constructs. Select the appropriate cloning backbone vector to insert theannealed oligos ( see Notes 2 and 4 ). Mix reagents listed below toset up the ligation reaction. Mix the same reagents without addingthe sgRNA oligo duplex to prepare a negative control reaction. Incubate the ligation reaction in a thermocycler followingthe parameters below. 3. (Optional but highly recommended) Plasmid-Safe treatment. Add Plasmid-Safe buffer, ATP, and the ATP-dependent Plasmid-Safe exonuclease as listed below to treat the ligation reaction andthe negative control to increase the effi ciency of cloning bydegrading unligated DNA fragments in the product mixture. Incubate the reaction mixture in a thermocycler followingthe parameters below. 4. Transformation. Add 2 μl of each reaction from previous step (including thenegative control ligation reaction) into chosen competentcells, such as Stbl3, transform following the manufacturer’sprotocol. Plate the transformed cells on Ampicillin selection LB agarplates or other type of plates depending on the selection markerof the backbone vector. 5. Plasmid preparation. Check the plate for presence of bacterial colonies after overnightincubation. Usually there should be no or less than fi vecolonies on the negative control plate, whereas over tens tohundreds of colonies will grow on the cloning plate, indicatinghigh cloning effi ciency. Pick two to three colonies from each agar plate and set upa small volume (typically 5 ml) LB culture for miniprep. Incubate and shake under 37 °C overnight, prepare plasmidDNA using a spin miniprep kit according to manufacturer’sprotocol. 6. (Optional) Digest the plasmid DNA to verify the insertion ofthe oligos. Digest the miniprep DNA with diagnostic restriction enzymes,BbsI and AgeI, if using the vector backbone PX330/PX335. Run the digested product on a 1 % agarose gel to visualize theband pattern of the digested product. When a lot of cloning is done at the same time, it is possibleto screen for correct insertion of the target sequence oligosby this digestion because a successful insertion will destroy theBbsI sites. After double digestion, clones with insertion of annealed sgRNA oligos will show only linearized plasmid,whereas clones without insertion will yield two fragments, withsizes of ~980 and ~7,520 bp in the case of pX330 when runningand visualizing on an agarose gel. 7. Sequencing verifi cation of positive clones. Sequence the clones with a forward primer that binds the humanU6 promoter to verify the successful insertion of guide sequences. The verifi ed clones can be then prepared using a Midi or Maxiplasmid extraction kit for downstream experiments. 3.2 Mammalian Cell Culture and Transfection As a general protocol, the steps below use HEK 293FT cells as anexemplar system to demonstrate CRISPR-Cas9 genome engineering. Additional suggestions can be found in Subheading 4 ( see Note 5 ). 1. HEK 293FT cell maintenance. Maintain the HEK cells in DMEM medium supplementedwith 10 % FBS (D10 medium) in an incubator at 37 °C temperatureand supplemented with 5 % CO 2 as recommended bythe manufacturer. Feed the cells every other day, and passagethe cells so they never reach over 75 % confluence. 2. Preparing cells for transfection. At 16–24 h before transfection, dislodge and disassociate thecells by trypsinization. Plate cells into 24-well plates containing500 μl antibiotic free medium at a density of 250,000 cellsper mL of culture medium, or 125,000 cells per well. Scale upor down the culture volume proportionally to the plate formatbased on the number of cells needed in the experiment. At thetime of transfection, the cells should be at around 75–85 %confl uence. 3. Transfection. Mix a total of up to 600 ng DNA containing the targetingconstructs in a microcentrifuge tube for each well of transfectionon a 24-well plate. For transfecting plasmid derived frompX330 or pX335, where a guide sequence has been clonedinto the backbone vector, directly use 600 ng of the preparedplasmid in the transfection mixture. When transfecting morethan one construct for alternative designs, e.g., the doublenickase method, or when multiplex genome cleavage is desired,mix equal molar ratio of all constructs to a total of 600 ng inthe transfection mixture ( see Notes 2 and 4 ). Add 50 μlOptiMEM medium OptiMEM medium to the DNA mixture. Mix well and spin down. Dilute Lipofectamine 2000 transfection reagent by adding1.5 μl of the reagent into 50 μl of OptiMEM, Mix well andincubate at room temperature for 5 min. Within 15 min ofdiluting the transfection reagent, add all the dilutedLipofectamine 2000 reagent into the DNA-OptiMEM mixtureprepared earlier. Mix well and spin down. Incubate themixture for another 20 min to allow the formation of DNA–Lipofectamine complex. Add the fi nal complex directly ontothe culture medium of each well on the 24-well plate. (Notrequired but highly recommended) Include a transfection controlto monitor transfection effi ciency. Use a control plasmidexpressing a fl uorescent protein, such as pCMV-EGFP, andtransfect this plasmid following same protocol as above intothe cell. Transfect another well of cells using the SpCas9 backbonevector, e.g. pX330 or pX335, as a negative control fordownstream processing and validation of assay. Replace the medium with warmed fresh medium around12–24 h post transfection. Maintain the cells for another48–72 h to allow suffi cient time for genome engineering mediatedby CRISPR-Cas9 system. 3.3 Genome Cleavage Analysis 1. Genomic DNA extraction. Extract genomic DNA from transfected cells using theQuickExtract DNA extraction kit following the manufacturer’srecommended protocol. Briefl y, disassociate cells from theplate, harvest by spin down the cell suspension at 250 × g for 5 min. Wash cell pellet with 500 μl of PBS and then resuspendedin QuickExtract solution. We typically use 50 μl for one well ina 24-well plate (scale up and down accordingly). Vortex thesuspension and incubate at 65 °C for 15 min, 68 °C for 15 min,and 98 °C for 10 min. 2. SURVEYOR assay to detect genomic cleavage. Use SURVEYOR assay to detect genomic cleavage after extractionof genomic DNA ( see Note 6 ), following the stepwiseinstructions provided in the SURVEYOR Mutation DetectionKit manual. A brief description of the steps is listed below. (a) Amplify extracted genomic DNA with a pair of primersdesigned for the target region of interest using a highfidelity enzyme such as Herculase II fusion polymerase ofKapa Hifi Hotstart polymerase. Typically an amplicon sizeof less than 1,000 bp is preferred as shorter amplicon givesmore specifi c amplifi cation products. (b) Visualize the PCR product on an agarose gel to check thespecifi city of the amplifi cation. It is important to havevery specifi c amplifi cation of genomic region to yieldaccurate SURVEYOR assay results as nonspecifi c bandswill interfere with the interpretation of gel electrophoresisanalysis. (c) Purify and quantify the PCR products, set up a denaturing/re-annealing reaction by mixing up to 400 ng of PCRproducts with water and re-annealing buffer (we typicallyuse the PCR reaction buffer and add to a fi nal concentrationof 1×, refer to the SURVEYOR assay manual for moreinformation). (d) Run the reaction in a thermocycler with followingparameters. (e) Digest the re-annealed products with SURVEYOR enzymekit at 42 °C for 1 h as recommended by the manufacturer’sprotocol. (f) Visualize the digested product using gel electrophoresis. For visualization of SURVEYOR assay results, we recommendedloading of the Surveyor Nuclease digestion productswith Polyacrylamide gel electrophoresis (PAGE) methodas it gives better solution compared with agarose gels. Quantifi cation of the assay results and the method toconvert it to an estimation of the frequency of indelsgenerated by CRISPR-Cas system in the population ofcells are described in Subheading 4 ( see Note 7 ). 3.4 Implementation of Homologous Recombination (HR) Using CRISPR-Cas System The guideline and considerations for designing a HR experimentto use CRISPR-Cas system to precisely modify the genomicsequence of interest by inserting, deleting, or replacing part of thegenome are described in Subheading 4 ( see Note 8 ). Briefl y, followingdesign and cloning of the HR template, perform HRexperiment following steps below. 1. HEK 293FT cell maintenance and the preparation of cells fortransfection. This part is same as the corresponding steps in Subheading 3.2 . Briefl y, plate cells into 24-well plates and make sure at the timeof transfection, the cells should be at around 75–85 %confl uence. 2. Transfection. Mix a total of up to 800 ng DNA containing the targeting constructsand the HR template vector (or single-stranded DNAoligos, see Note 9 ) in a microcentrifuge tube for each well oftransfection on a 24-well plate. Generally, apply a molar ratio of1:3–5:1 for the targeting vector and HR template vector. (Optional but recommended) Titrate different molar ratiobetween the targeting vector and HR template vector to test theoptimal condition for the HR experiment. Add 50 μl OptiMEMmedium to the DNA mixture. Mix well and spin down. Dilute Lipofectamine 2000 transfection reagent by adding2 μl of the reagent into 50 μl of OptiMEM, Mix well and incubateat room temperature for 5 min. Within 15 min of dilutingthe transfection reagent, add all the diluted Lipofectamine2000 reagent into the DNA-OptiMEM mixture prepared earlier. Mix well and spin down. Incubate the mixture for another20 min to allow the formation of DNA–Lipofectamine complex. Add the fi nal complex directly onto the culture mediumof each well on the 24-well plate. Transfect another well of cells using the SpCas9 backbonevector, e.g., pX330 or pX335, together with the same amountof HR template vector as a negative control. Replace the medium with warmed fresh medium around12–24 h post transfection. Maintain the cells for another 72 hto allow suffi cient time for HR mediated by CRISPR-Cas9 systemand the template. 3.5 Verifi cation and Quantifi cation of HR Effi ciency To verify the homologous recombination between the HR templateand the endogenous genome, restriction fragment lengthpolymorphism (RFLP) assay for HR can be applied. 1. Genomic DNA extraction. Extract the genomic DNA using the same extraction protocolusing the QuickExtract DNA extraction kit as in theSURVEYOR assay (Subheading 3.3 ). 2. Target region amplifi cation. Amplify the genomic region of interest by a HR testing primerset where the two primers bind outside the homology regionto avoid false positive results given by amplifi cation of the residueHR template. 3. Perform RFLP digestion. Run the resulting PCR product on an agarose gel to check forspecifi city of amplifi cation, as nonspecifi c PCR products willinterfere with the assay and prevent accurate quantifi cation ofHR effi ciency. In many cases, several pairs of HR testing primersets should be screened to obtain robust, specifi c amplicons. Purify the PCR amplifi cation product by standard PCRpurifi cation, or in the case where clean PCR product cannot beobtained, gel extract the desired amplicon following separationof PCR product on an agarose gel. Digest the purifi ed products with the appropriate enzymecorresponding to the design of the HR template ( see Note 8 ),and visualize on an agarose gel or PAGE gel. The latter usuallygives better resolution and is highly recommended. The effi -ciency of HR in the population of cells assayed can be estimatedby the following formula: HR percentage (%) = ( m + n / m + n + p ) × 100 Here the number “ m ” and “ n ” indicate the relative quantityof bands from digested genomic PCR products, whereasthe “ p ” equals the relative quantity of undigested products. 4. Perform additional Sanger and next-generation DNA sequencingto verify the presence of desired engineered sequenceswithin the genome. Briefl y, clone the genomic PCR productinto a sequencing vector, TOPO-TA, or other blunt-endcloning method, and perform Sanger sequencing to detectrecombined genomic amplicons. Alternatively for higherthroughput, subject the genomic PCR products to next-generationsequencing. 4 Notes 1. Identifi cation and selection of target genomic site. The twoprimary rules for identifying a target site for the SpCas9 systemare: (1) fi nding the “NGG” PAM sequence which is requiredfor SpCas9 targeting, and (2) picking a sequence of 20 bp inlength upstream of the PAM to its 5′ end as the guide sequence. Following these two guidelines, multiple potential target sites can be usually found within the genomic region of interest(Fig. 2 ). Additionally, when using U6 promoter to expresssgRNA (such as pX330, pX335), we suggest adding the G (notreplacing but add one more base) because the human U6 promoterrequires a “G” at the transcription start site to havehighest level of expression (Fig. 2 ). While we do notice thatsometimes the sgRNA will still work without the extra “G”, itis generally better to have this additional base. In the casewhere the guide sequence starts with a base “G”, this additioncan be omitted. In our open-source online resources website( http://crispr.genome-engineering.org ), we provide the mostup-to- date information for using the CRISPR-Cas9 system forgenome engineering, focusing on the SpCas9 system. Additionally, we also developed an online tool for the selectionof SpCas9 targets for different organisms including human,mouse, zebrafi sh, C. elegans , etc. This tool can greatly facilitateand simplify the process of performing target selection in batch( http://crispr.mit.edu/ ). Because the effi ciency of differenttargets could vary considerably depending on the guidesequences, we highly recommend testing multiple target sitesfor each gene or region of interest and selecting the most effectivetarget ( see Note 1 ). In the case of double nickase design,we recommend individually testing each target with the wildtype SpCas9 system to assess the cleavage effi ciency of individualguides and then combine the most effi cient pair ofguides with opposite directionality and appropriate spacing forthe genome engineering application. 2. Design of oligos for inserting guide sequences into backbonevectors. The cloning vectors we use for typical SpCas9 genometargeting are pX330 for the wild type SpCas9 and pX335 forthe nickase version SpCas9n. Both vectors are mammaliandual- expression vectors, which enables the co-expression ofSpCas9 protein driven by the potent constitutive promoterCBh and sgRNA driven by the RNA Pol III human U6 promoterin mammalian cells (Fig. 3 ). CBh promoter is a hybridpromoter derived from the CAG promoter, which have beenvalidated to support strong expression of transgene in multiplecell types/lines, including HEK 293FT, mouse Neuro-2a,mouse Hepa1-6, HepG2, HeLa, human ESCs, and mouseESCs. To clone custom guide sequences into these backbonevectors, a pair of oligos encoding the guide sequences can beordered with the appropriate overhangs (Fig. 3 ), then annealedto form a clone-ready duplex DNA fragment. The vector canbe then digested using BbsI, and a pair of annealed oligos canbe cloned into the backbone to express the correspondingsgRNA (Fig. 3 ). The oligos are designed based on the targetsite sequence selected in previous section. A common confusionsometimes in cloning the guide into backbone vector is to include the “NGG” PAM sequence in the guide sequence. Hence, it is important to check that only the 20 bp sequenceto the 5′ end of the PAM is being used for designing the oligosequences to order. An alternative way of designing oligos fordirectly amplifying a PCR fragment that contains the U6 promoterdriving a sgRNA could also be employed for testing theguide sequences, which simplifi es the test by avoiding the needof cloning, but might be less effi cient than using the clonedvector plasmids ( see Note 4 , ). Fig. 3 Schematics for the cloning backbone vectors pX330/pX335 with oligo design for inserting guidesequences. The pX330 and pX335 vectors contains dual-expression cassettes for both the SaCas9 protein andthe sgRNA. Digestion of the backbone with Bbs I Type II restriction sites ( blue ) generates the complementarycloning overhangs to the annealed oligos ( purple boxed ). Note that a G–C base pair is added at the 5′ end ofthe guide sequence for optimal U6 transcription. The oligos contain overhangs for ligation into the overhangsof Bbs I sites. The top and bottom strand orientations is exactly identical to those of the genomic target butexclude the “NGG” PAM. Parts of this fi gure are adapted from 3. Screening of multiple guides. For most applications, we screenfor at least three guide sequences within the target genomicregion in an effort to fi nd the most effi cient ones. This isbecause while CRISPR-Cas9 system works very effi ciently, theactual cleavage effi ciency could be affected by the sequence ofthe guide, the accessibility of local chromatin, the activity ofthe endogenous DNA repair pathways, and other guide-specific or cell-type-specifi c factors. Hence, to ensure that a validguide sequence is obtained, this screening process is highlyrecommended. Following the same logic, a guide sequencethat has been verifi ed in one cell type will not necessarily workto the same effi ciency in another cell type or condition. Hence,additional optimization or re-screening of new guide sequencesmight be required when moving from one experimental sys-tem to another. This same situation is also applicable to theHR experiment where the HDR effi ciency can very considerablyamong different types of cells or tissues. 4. Additional strategy for screening guides and backbones for differentapplications. We have also developed another way ofquickly screening guide sequences with amplifi ed PCR products. In this design, two primers are used to amplify the U6RNA-expression promoter, where the forward primer binds tothe 5′ beginning of U6 promoter, and the reverse primer bindsto the 3′ end of the U6 promoter. Because the reverse primeralso contains a long extension that can add on the guidesequence and the chimeric sgRNA scaffold, the amplifi ed PCRproduct contains all necessary elements for expressing a sgRNAcontaining the guide specifi ed in the reverse primer. Hence,the screening of guide sequences can be done by co-transfectingthis PCR product with a backbone vector expressing theSaCas9 protein. Because many application of CRISPR-Cas9genome engineering involve cell lines that might be diffi cult towork with, e.g., cell lines that are hard to transfect, we developedadditional backbone vectors to facilitate selection andscreening for transfected cells. These vectors contain the fl uorescentmaker protein, GFP, or the selectable puromycin resistancegene, linked to the expressing of SaCas9 via a 2A peptidelinker. These constructs will enable fl uorescence activated cellsorting (FACS) or the selection of transfected population,which can further improve the overall effi ciency of genomeengineering particularly in the case of HR applications. Additional details on these designs and backbones can befound in our recent publication . 5. Cell line choice for validation of guide design. Functional validationof targeting constructs bearing the designed guides canbe carried out in relevant cell lines, e.g., HEK 293FT, K562,Hela for human genome engineering, or Neuro-2a, Hepa1-6for mouse. This process takes advantage of some favorableexperimental properties of these lines, such as robust and easymaintenance, effi cient transfection, etc., before embarking oncomplicated procedures in other mammalian systems. Nonetheless, achieving best results for each experiment mightrequire additional optimization ( see Note 2 ). Moreover, dueto the genetic and epigenetic differences between cell types orsubjects of study, results obtained from one cell type might notnecessarily correspond to those from another cell type of thesame species ( see Note 2 ). 6. Mechanism of SURVEYOR nuclease assay. Following thedelivery of SpCas9 and the sgRNAs into mammalian cells, theinduced genomic cleavage could be assayed by the SURVEYORassay, which could detect modifi cation of genomic DNA within of the cells will be modifi ed by SpCas9 so that their genomicsequence at target site is different from the un-modifi ed population. Hence, in the assay, it is possible to amplify region ofinterest from genomic DNA via PCR, then through a denaturingand re-annealing process to form mismatched DNA. Thismismatched DNA can then be recognized by the SURVEYORnuclease and cleaved for visualization on analytical gels. Toquantify the effi ciency of genomic cleavage, one can then assessthe percentage of cleaved products as a surrogate for the percentageof indels generated within the target genomic region. 7. Analysis of SURVEYOR assays results. To calculate the genomecleavage effi ciency of a tested target, quantify the band intensityof SURVEYOR assay products visualized by PAGE usingthe following formula: Indel percentage (%) = (1 . √(1 . x )) × 100, where x = ( a + b )/( a + b + c ) In this formula, the number “ a ” and “ b ” represent therelative quantities of the cleaved bands, while “ c ” equals to therelative quantity of the non-cut full-length PCR product. Other methodology of detecting the genomic cleavage canalso be applied. One such method is to clone the SURVEYORPCR products into a sequencing vector, e.g., pUC19, andtransformed into E. coli . These individual clones can be thensequenced via Sanger sequencing to reveal the identity ofgenome modifi cations. Additionally, the percentage of modified clones can also be used as a measurement for the effi ciencyof genome engineering. Alternatively, the PCR products couldalso be sequenced in a more high-throughput way with nextgenerationsequencing. 8. Design and synthesis of repair template for HR experiment. Forintroducing a precise genomic modifi cation into the genome,the HDR pathway can be employed. This is achieved by cotransfectingSpCas9 constructs (derived from pX330 or pX335)bearing guide sequences with a HR template in the target cellline. After recombination, modifi cations such as point mutation,small and large insertions/deletions, or other type ofchromosomal changes could be engineered into the endogenousgenome. A few considerations for the choice of guide: (a) Typically, a screening for the most effi cient guide sequenceis performed fi rst. We recommend picking several (three tosix) targets within the genomic region of interest followingprotocols listed earlier. Tests are then performed to assaythe cleavage effi ciency of each of these guides. Then, theactual HR experiments can be carried out with themost effi cient guides (also see additional considerations inNotes 3 and 4 ). (b) For maximize the effi ciency of HR, it is recommended thatthe cleavage site of the guide is as close to the junction ofthe homology arm, i.e., the size at which genome modifi -cations are introduced, as possible. Usually this distanceshould be less than 100 bp, ideally less than 10 bp. (c) To minimize the off-target cleavage, the double nickasedesign can be used. In this case, multiple guide sequencescan be fi rst tested individually, and typically the combinationof highest cutting guide designs with appropriatedirectionality will yield highest cleavage when used in thepaired fashion, thus giving best results in HR experiment. 9. The HR template is essentially the desired sequence that needsto be present in the engineered genome, fl anked by twohomology arms bearing the same sequence as the referencegenome. Below are considerations for the choice of HRtemplate: (a) It is usually advised to insert a testable marker in the HRtemplate to facilitate the assay for successful HR events. For example, a restriction site could be inserted to allowRFLP assay. Alternatively, the insertion of fl uorescent proteinsor selectable drug-resistance genes such as puromycin-resistance cassette can also be used. (b) For introducing single-point mutation the best HR templatefor transfection is usually single-stranded DNA(ssDNA) oligos. For ssDNA oligo design, we typically usearound 50–90 bp homology arms on each side and introduceyour mutation/modifi cation in between the twoarms. When ordering long oligos, ultramer oligo (IDT) isrecommended. (c) For introducing larger genomic modifi cation, plasmidDNA vector can be used because of the length limit ofssDNA oligos. When designing a plasmid-based HR template,a minimum of 800 bp homology arms on each sideis recommended. (d) If you have intact “protospacer + PAM” sequence within theHR template, it can lead to the HR template being degradedby Cas9. Hence, it is recommended to make silent mutationsto destroy the sgRNA-binding site, or avoid putting inthe full target site in the HR template by choosing targetsites that span the site of modifi cation. For making silentmutations, one good option is to mutate the PAM “NGG”within the HR template, as the PAM is required for cleavage. For example, change the “NGG” to “NGT” or“NGC”, in addition to mutations in the spacer itself, couldusually prevent degradation of donor plasmid. 本文转自丁香园: 查看原文
新型 DNA 编辑技术 CRISPR 从 2013 年至今都是很热门的领域。 DNA 编辑就是改变生物体内已有的基因,这个概念在遗传学领域早就有了,并不是新名词。 CRISPR 的影响力之所以大,是因为它使得 DNA 编辑的效率大大提高,使得原来一些耗时低效的遗传学实验变得简单,甚至使一些不可能的实验变为可能。崔健说:不是我不明白,是这世界变化快。这就是 CRISPR 带来的改变。 在 CRISPR 产生之前,要改变生物体内的基因主要通过 DNA 同源重组的方法。当两段高度相似的 DNA 序列相遇时,它们可以相互结合然后交换序列,这个过程被称为 DNA 同源重组。通过这个过程,可以把做了改变的 DNA 片段替换生物体内原有的 DNA 。 DNA 重组率高,说明这个物种的 DNA 容易被改变,敲除或改变基因在这个物种中也容易被实现。 虽然还没有直接的证据,但生物体 DNA 被改变的容易程度与其染色体的复杂程度紧密相关。染色体的复杂程度主要取决于 DNA 所缠绕的组蛋白的种类、修饰的多样性以及相互作用。例如,病毒与细菌的 DNA 没有被组蛋白包裹,它们的 DNA 重组率很高。酵母与老鼠都有组蛋白,但酵母的 DNA 重组率比老鼠要高很多。所以,通过 DNA 同源重组的方法在酵母中敲除一个基因比在老鼠中要容易得多得多得多。高等动物拥有复杂而且难以被入侵的染色体结构是符合信息安全的逻辑的:越庞大重要的信息需要越安全的保护。 CRISPR 把一套真核生物没有的 DNA 编辑系统引进来,轻而易举地突破高等动物的染色体防线,让高等动物的基因敲除或者替换变得容易很多。当然,在没有 CRISPR 之前也有别的 DNA 编辑方法,例如 zinc finger nuclease 和 TALEN ,这些方法比 CRISPR 要复杂一些而且限制也多一些。 由于 CRISPR 的高效率,科学医学界的一大呼声是希望 CRISPR 能用于修复人体内的某些基因缺陷,用于治疗艾滋病,先天性遗传病,甚至是癌症。但最近的一些研究表明, CRISPR 的专一性并没有原来预测的那么好,因为人的基因组很大, CRISPR 会非特异性地改变一些正常的基因。所以,实现这些愿望可能会有些漫长,也有可能会被证明不可能。 同时,由于 CRISPR 的高效率,有警惕的科研人员都很明白 CRISPR 可能会带来的灾害。现在的一个比较强烈的呼声是要限制 CRISPR 在人体身上的应用,原因很简单:除了副作用的不确定性,还有伦理宗教的问题。另一方面就是要防止基于 CRISPR 的新型病毒或纳米颗粒给人类社会带难以预测的灾难。与后者有关的声音目前比较弱小,可能是这牵涉到国防政治的问题,不便摆到桌面上来说。 CRISPR 的威力在那里,就看人类怎么用它。
CRISPR 先驱者荣获 2015 生命科学突破奖 诸平 据《 科学家 》( The Scientist )网站 2014 年 11 月 18 日 报道, 2015 生命科学突破奖 ,被 CRISPR 的先驱者荣获。下面的3人照片从左到右依次是 艾曼纽·卡彭特( Emmanuelle Charpentier )、詹妮弗·杜德纳( Jennifer Doudna )以及 维克托·安布罗斯( Victor Ambros )。 Alim-Louis Benabid en 2013 Gary Ruvkun 2015 年生命科学突破奖确认了六位科学家,艾曼纽·卡彭特( Emmanuelle Charpentier )、詹妮弗·杜德纳( Jennifer Doudna )、 维克托·安布罗斯( Victor Ambros ) 、阿里姆·路易斯·本纳比德( Alim Louis Benabid )、加里·鲁夫昆( Gary Ruvkun )以及 C. 戴维·阿里斯( C. David Allis ),其成就在于基因编辑、基因调控和帕金森氏症治疗方面的突出贡献。 300 万美元的奖项是由一群包括俄罗斯企业家尤里·米尔纳( Yuri Milner )及其夫人茱莉雅·米尔纳( Julia Milner )、脸书( Facebook )的创办人马克 • 扎克伯格 ( Mark Zuckerberg ) 及其夫人普莉希拉·陈( Priscilla Chan )、 谷歌公司的创始人谢尔盖 • 布林 ( Sergey Brin ) 和安妮 · 沃西基( Anne Wojcicki ) , 马云夫妇( Jack Ma and Cathy Zhang )等科技亿万富翁提供的。该奖项于 2014 年 11 月 9 日 在美国加利福尼亚的山景城举行了电视授奖仪式。由亿万富翁企业家共同设立的“科学突破奖”旨在奖励在生命科学等领域取得重要成就的科学家,给他们提供更自由和更多的机会,帮助他们取得更大的成就。每年的获得者将加入评选委员会,参与下一届获奖者的评选。 自 2013 年这一奖项首次颁发以来,已有多位科学家荣获了巨额奖金,最新的突破奖在物理学和生命科学外还增加了数学奖项,尤里·米尔纳本人表示我们的世界总是围绕体育和娱乐名人转。但科学名人可能进入不了前 200 或 300 名,这种排名结果与其贡献相比,极不对称。 本次生命科学突破奖获得者,德国亥姆霍兹感染研究中心( HelmholtzCenter for Infection Research in Germany )和瑞典于默奥大学( Umeå University in Sweden )的艾曼纽·卡彭特( Emmanuelle Charpentier )及美国加州大学伯克利分校( University of California, Berkeley )詹妮弗·杜德纳( Jennifer Doudna ),因为他们发现了 CRISPR 的细节而获奖 , CRISPR 是一种被广泛应用于基因工程的细菌抗病毒系统。另外还有法国约瑟夫·傅里叶大学( Joseph Fourier University in France )的神经外科医生阿里姆·路易斯·本纳比德,是因为将 深层脑部刺激术( deep-brain stimulation ) 作为帕金森症一种治疗方法而获奖;而美国麻省大学医学院( University of Massachusetts Medical School )的维克托·安布罗斯( VictorAmbros )和美国哈佛医学院( Harvard Medical School )的加里·鲁夫昆( Gary Ruvkun )是因为他们对 miRNAs ( microRNAs )调控基因研究的突出贡献而获奖。美国洛克菲勒大学( Rockefeller University )的 C. 戴维·阿里斯( C. David Allis )获奖原因,是因为特别在表观遗传学( epigenetics )、组蛋白( histone proteins )的共价修饰蛋白对基因调控和疾病有广泛影响方面的研究。 詹妮弗·杜德纳告诉《卫报》 ( The Guardian )说, 她得到获奖的信息,使她 “ 激动和震惊 , 同时也非常兴奋。我们的研究成果是在 2012 年发表了的,但是谁也没有预料到,它竟然会有如此的变革力。” CRISPR-Cas9 系统已经被证明是一种强大的研究工具 , 并显示有望作为一种基因治疗的方法, 治疗囊肿性纤维化( cystic fibrosis )和血液疾病( blood disorders )。 洛克菲勒大学的 C. 戴维·阿里斯( C. David Allis )告诉《卫报》说: “ 当你决定追求生命科学领域一个非常基本的问题如基因调控时 , 对于得到一个引人注目的突破奖从来就不抱任何希望。它显示了将科学研究作为一种事业是多么美妙绝伦。 ” 这种精神境界是何等的高尚和值得可敬,与那些魂牵梦绕,无时无刻不再想如何快步直奔领奖台的唯利是图者相比较,可以说有天壤之别!科学研究的真谛是探索未知,而不是获得何种奖项。更多阿里斯的信息请浏览 ( NASprofile.pdf )。 CRISPR 基因组编程技术是近两年兴起的一种新技术, 2013 年,两篇 Science 新闻开创了 CRISPR 基因组编辑技术的新时代,随后生命科学界刮起了 CRISPR 风暴,迄今为止 CRISPR 方法已迅速席卷了整个动物王国,成为 DNA 突变和编辑的一种重要技术。 CRISPR 全称为 clustered regularly interspersed short palindromic repeats ,是源于细菌及古细菌中的一种后天免疫系统,它可利用靶位点特异性的 RNA 指导 Cas 蛋白对靶位点序列进行修饰。直到今年,科学家们才开始利用这一系统在活体动物基因组中生成靶向突变,删除原有的基因或插入新基因。对于这一技术詹妮弗·杜德纳表示,“修改生物体基因特定部分的能力,对于增加我们对生物体的认知是必不可少的。这是该领域的一种巨大跨越,因为它意味着基本上任何人都能够使用这种基因编辑或者重新编写的技术带给哺乳动物基因变化。” 与过去数十年里进行基因工程的其它任何方法相比, CRISPR 技术的优点就在于它使用的是单一的酶。这种酶不需要改变你设定目标的每一个点,你只需要使用一个不同的 RNA 副本对它进行重新编辑,这很容易设计和实现。生命科学突破奖获得者中有几位科学家则分别是在遗传学, RNAi 等领域的突出贡献而获奖,如哈佛医学院遗传学教授加里·鲁夫昆 就是一位微小 RNA(miRNA) 研究领域的著名科学家,他曾发现了首例微小 RNA : lin-4 通过与目标信使 RNA 不完全碱基配对,来调控这些目标的翻译的机制,并发现了第二个微小 RNA —— let-7 ,以及它在动物(包括人类)系统发育中如何保护的。 2008 年获 拉斯克基础医学奖 ( Albert Lasker Basic Medical Research Award )。 miRNA 是一类非编码小 RNA ,其长度为 19 到 25 个核苷酸,在真核生物的多种发育和生理过程中发挥着重要的调节作用。加里·鲁夫昆等人就曾通过分析比较了 86 个不同真核基因组序列,分析了小 RNA 辅助因子的系统发生谱,并指出,在 RNA 剪接和小 RNA 介导的基因沉默之间存在密切关联。 The 2015 Breakthrough Prizes in Life Sciences The Breakthrough Prize in Life Sciences honors transformative advances toward understanding living systems and extending human life, with one prize dedicated to work that contributes to the understanding of Parkinson’s disease. Alim Louis Benabid , Joseph Fourier University, for the discovery and pioneering work on the development of high-frequency deep brain stimulation (DBS), which has revolutionized the treatment of Parkinson’s disease. C. David Allis , The Rockefeller University, for the discovery of covalent modifications of histone proteins and their critical roles in the regulation of gene expression and chromatin organization, advancing the understanding of diseases ranging from birth defects to cancer. Victor Ambros , University of Massachusetts Medical School, and Gary Ruvkun , Massachusetts General Hospital and Harvard Medical School, for the discovery of a new world of genetic regulation by microRNAs, a class of tiny RNA molecules that inhibit translation or destabilize complementary mRNA targets. Each received a $3 million award. Jennifer Doudna , University of California, Berkeley, Howard Hughes Medical Institute and Lawrence Berkeley National Laboratory, and Emmanuelle Charpentier , Helmholtz Center for Infection Research and Umeå University, for harnessing an ancient mechanism of bacterial immunity into a powerful and general technology for editing genomes, with wide-ranging implications across biology and medicine. Each received a $3 million award. 更多信息请浏览: https://breakthroughprize.org/?controller=Pageaction=newsnews_id=21
After transformation of the CRISPR/Cas9 constructs in rice callus and regeneration of transgenic plants, many Cas9-positive T0 plants were identified for each of the targets and these plants were analysed to detect mutations in the targeted sequence regions (Table 1 ). DSB must have happened in nearly half of the T0 plants tested because 44.4% of them carried mutations. Mutation rate varied in a wide range from target gene to target gene, that is, from 21.1% to 66.7%. For the two target sites in the same OsYSA gene, the difference was relatively small (43.1% and 66.7%). The CRISPR_Cas9 system produces specific and homozygous targeted gene editing in.pdf
没有人愿意两次掉进同一个陷阱,同样,生物体也进化出了各种机制防治被同一种病毒感染两次。人体以及其他高等生物产生的抗体能够记住他们感染过的病毒,从而防止第二次感染。最近三年来,研究人员惊奇的发现,单细胞的细菌也有类似的 ” 记忆性 ” 免疫叫做 ”CRISPR” 。这个故事起源于 2007 年,有一家出售用于生产酸奶的细菌的农业科技公司,客户反馈说他们收到的部分细菌感染了病毒,不能进行酸奶发酵。于是这家公司派出一个研究小组去调查此事。研究员同时对两类酸奶细菌做 DNA 测序。第一类是正常的对病毒免疫的好细菌,第二类是容易被病毒感染的不好的细菌。他们发现这两类细菌 DNA 唯一不同的是,在第一类细菌的 DNA 上多出了一小段,而且有意思的是这一小段 DNA 竟然和他们免疫的病毒完全吻合。后来一系列实验证明,就是这一小段 DNA ,就像是一个标签,让细菌记住了这个病毒。 这个实验发表之后引起了极大的轰动,由于是一个全新的领域,很多的问题暂时无法理解。但大概的思路是这样的。当病毒入侵进入细菌时,细菌的一个蛋白复合体能够截取一小段病毒的 DNA ,大约 20 个碱基 . 这一小段 DNA 能够镶嵌在细菌自己的基因组上 , 并在细菌复制时传给下一代。当同样的病毒试图再次入侵此细菌时,这一小段 DNA 就会被转录成 RNA ,并且和外来的病毒 DNA 配对,如果碱基配对完全正确的话,细菌就会激活一个清除机制来清除此外来的病毒,从而防止被感染。 我是 2010 三月份时开始进入这个领域的。我们发现, CRISPR 具有更新换代的机制。简单说,细菌不可能一直不停的获取病毒 DNA 片段,并插入到自己的 DNA 中,这样的话细菌的 DNA 就会无限制的增长了。一个细菌平均只能保留 20 个病毒 DNA 片段,那些老一点的片段就会慢慢的从细菌的 DNA 中删掉。新的 DNA 片段源源不断的添加进来,保证 CRISPR 能提供最新的免疫。--Jiankui He and Michael Deem, Phys.Rev.Lett.105:198701 (2010) 关于 CRISPR 的研究还处于起始阶段,所有相关的文献不过 20 篇。但是这是一个全新的免疫的机制,它在细菌抗药性,细菌的基因转接等等方面有着巨大的潜在应用。免疫这一方向已经发了四个诺贝尔奖了,比如说我的合作者之一的 Andrew Fire 就因 RNAi 获奖, CRISPR 的发现很有可能再拿一个诺贝尔奖。 CRISPR
 标签: CRISPR
标签: CRISPR