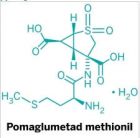
裸头草碱的 1 期临床试验未显示不良反应 诸平 Psilocybin is a naturally occurring psychedelic substance produced by psilocybin mushrooms, also called ‘magic’ mushrooms. 据“医学快报”( Medical Xpress ) 2019 年 12 月 13 日 提供的消息,英国伦敦国王学院( King's College London )和精神保健公司 Compass Pathways 的研究人员团队发布了 裸头草碱 (psilocybin ) 的 1 期临床试验结果。除了在美国神经心理药理学院( American College of Neuropsychopharmacology ) 年会上 ( annual meeting )宣布结果外,团队成员还与新闻界进行了交谈。他们 报告说 ( reported ),到目前为止,他们并未发现服用该药物的志愿者有不良反应。 裸头草碱是蘑菇中一种具有迷幻作用的成份,可有效降低癌症患者的焦虑情绪。但是,也有报道指出,裸头草碱具有积极和消极的身体和心理影响,该药可引发精神病发作 风险增加。目前,裸头草碱在美国还不能用于临床,因为它被美国药物管理局 (DEA) 列为一级药物。 在医疗机构中,医生已经测试过裸头草碱,用于治疗 丛集性头痛 ( cluster headaches ), 晚期癌症 焦虑症( end-stage cancer anxiety ), 抑郁症 ( depression )和其他焦虑症( other anxiety disorders )。尽管如此,还是有 科学家质疑裸头草碱作为治疗手段的有效性和安全性。 英国伦敦国王学院( King's College London )和精神保健公司 Compass Pathways 的研究人员团队近日发布裸头草碱 (psilocybin) 的 1 期临床试验结果: 1 期临床试验仅用于测试该药物的安全性 - 并未试图找出该药物是否可减轻抑郁症状。没有一个志愿者患有抑郁症。这项双盲试验包括为 89 名健康志愿者提供 10 mg 或 25 mg 剂量的药物(或安慰剂),并进行后续治疗。对每个志愿者进行长达 12 周的不良反应评估。 研究人员想知道,分组治疗与一对一治疗是否会对结果产生影响。他们指出,每个 志愿者都 被指派了一名治疗师,他们与志愿者一起参加了治疗试验。 Compass Pathways 的代表告诉媒体,他们有兴趣了解是否有可能简化治疗程序以加快临床试验过程。他们还报告说,试验期间共进行了 25 次给药。 研究人员报告说,任何志愿者都没有严重的不良反应。轻微的不良反应具有迷幻性质。他们还发现,志愿者没有任何认知功能或情绪上的不良影响。他们通过试验得出结论,表明裸头草碱作为治疗慢性抑郁症的药物结合 治疗 的可行性。国王学院的代表指出,他们的研究是迄今为止裸头草碱规模最大的研究,并称结果令人鼓舞。更多信息请注意浏览相关报道。 Psychedelic drug to be tested for treatment-resistant depression in Houston Active ingredient in magic mushrooms reduces anxiety and depression in cancer patients The potential of psilocybin to alleviate psychological distress in cancer patients is revealed Psilocybin: Safety, Side Effects Intriguing Research
世界卫生组织WHO 5月18号发布了一则消息:世界一些主要的医学科研基金会和非政府组织同意实行一项新的标准,要求其所资助或支持的所有临床试验都要注册并公开结果。不难想像,许多权威医学期刊也会跟进,对相关投稿论文做出相应的要求。以下为新闻节选: “Indian Council of Medical Research, the Norwegian Research Council, the UK Medical Research Council, Médecins Sans Frontières and Epicentre (its research arm), PATH, the Coalition for Epidemic Preparedness Innovations (CEPI), Institut Pasteur, the Bill Melinda Gates Foundation, and the Wellcome Trust等联合签署声明,同意在接下来的12个月内实行新的标准,要求其资助或支持的临床试验都要在公众可访问的网站注册,并在指定的时间内公开试验结果。 一些研究结果表明,今天约有50%的临床试验因为结果阴性而没有得到公布,这使得关于疫苗、药品和医疗器械的风险与益处的研究文献不完整甚至误导,导致次优甚至有害的产品。 2015年,WHO就基于世界医学组织(WMA) 2013赫尔新基宣言发布了其要求临床试验公开结果的声明,定义了公开结果的时间框架,并号召未发表的临床试验也要公开结果。今天由以上各主要基金和组织签署的协议意味着WHO和WMA声明中的伦理原则将会在每年成千上万的临床试验中得到加强。” 请点击“阅读原文”访问WHO新闻英文原文。 题图:坏血病的克星,作者Robert A Thom. 描绘了世界临床试验的鼻祖苏格兰医生James Lind于1747年开展的柑橘类水果与其他水果治疗坏血病的对照试验。 (图片来源https://research.ncl.ac.uk) ORCID上海研讨会将于2017年6月19号于上海交通大学举办, 请访问 http://lxi.me/64rcw 或扫描二维码查看详情或在线报名
Lancet Neurol——AD第一个tau疫苗临床试验显示出良好的安全性 Title: Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease : a randomised , double-blind , placebo-controlled , phase 1 trial . Authors: Novak et al. Journal: Lancet Neurol. 2016 Dec 9. Absta Abstract BACKGROUND: Neurofibrillary pathology composed of tau protein is a main correlate of cognitive impairment in patients with Alzheimer's disease . Immunotherapy targeting pathological tau proteins is therefore a promising strategy for disease -modifying treatment of Alzheimer's disease . We have developed an active vaccine , AADvac1 , against pathological tau proteins and assessed it in a phase 1 trial . METHODS: We did a first-in-man, phase 1 , 12 week, randomised , double-blind , placebo-controlled study of AADvac1 with a 12 week open-label extension in patients aged 50-85 years with mild-to-moderate Alzheimer's disease at four centres in Austria. We randomly assigned patients with a computer-generated sequence in a 4: 1 ratio overall to receive AADvac1 or placebo. They received three subcutaneous doses of AADvac1 or placebo from masked vaccine kits at monthly intervals, and then entered the open-label phase , in which all patients were allocated to AADvac1 treatment and received another three doses at monthly intervals. Patients , carers, and all involved with the trial were masked to treatment allocation. The primary endpoint was all-cause treatment-emergent adverse events, with separate analyses for injection site reactions and other adverse events. We include all patients who received at least one dose of AADvac1 in the safety assessment. Patients who had a positive IgG titre against the tau peptide component of AADvac1 at least once during the study were classified as responders. The first-in-man study is registered with EU Clinical Trials Register, number EudraCT 2012-003916-29, and ClinicalTrials.gov, number NCT01850238 ; the follow-up study, which is ongoing, is registered with EU Clinical Trials Register, number EudraCT 2013-004499-36, and ClinicalTrials.gov, number NCT02031198 . FINDINGS: This study was done between June 9, 2013, and March 26, 2015. 30 patients were randomly assigned in the double-blindphase : 24 patients to the AADvac1 group and six to the placebo group. A total of 30 patients received AADvac1 . Two patients withdrew because of serious adverse events. The most common adverse events were injection site reactions after administration (reported in 16 vaccinated patients ). No cases of meningoencephalitis or vasogenic oedema occurred after administration. One patient with pre-existing microhaemorrhages had newly occurring microhaemorrhages. Of 30 patients given AADvac1 , 29 developed an IgG immune response. A geometric mean IgG antibody titre of 1 :31415 was achieved. Baseline values of CD3+ CD4+ lymphocytes correlated with achieved antibody titres. INTERPRETATION: AADvac1 had a favourable safety profile and excellent immunogenicity in this first-in-man study. Further trials are needed to corroborate the safety assessment and to establish proof of clinical efficacy of AADvac1 . FUNDING: AXON Neuroscience SE.
A privately held start-up company, Syntellix, has announced the first Mg-alloy (WE43-based) screw for bone correction osteotomy in foot surgery. http://syntellix.com/en/news/ http://syntellix.com/en/products/ the latest clinical paper: http://www.biomedical-engineering-online.com/content/12/1/62
隔着 NIH几条街,联邦政府买单让我来学习对医学研究有益的技能,DoubleTree宾馆里看着争论,该相信谁呢?该相信郝炘所介绍的NIH官方回复,还是肖传国反驳?经常怀疑并不一定是坏事,而一味肯定一定不是什么好事。学习几个术语,可能更能加深理解临床试验的准则和要求。 第一:protocol,研究方案:这是临床试验的法定条文,所有的研究内容、实施方案都要体现在这个本本上,而该方案能否通过需要几层审批。最基本的是大学或医院的伦理委员会,简称(IRB)。如果是新药研究,即便是验证“老药新用”也必须获得FDA批发的IND;而显然肖氏手术的Nerve Rerouting Treatment for Neurogenic Bladder inSpina Bifida(NCT01096459)这一研究方案没有被NIDDK所采纳,而这一方案递交的时间应该是2013年1月,应该改进的、新的方案,这与此前批准实施的研究方案应该是不一样的,没有通过NIH及外部专家的评审,而这个方案没有通过的原因是: The experts thought that the proposed trial and itsprimary outcome measures were inadequately designed. 换一句话说,研究方案设计存在缺陷,而任何一个临床试验,只有一个关键,那就是:primary outcome,也就是解决唯一的、用一句话概括的关键问题。 第二:primary outcome,主要设定结果。开展临床试验的关键是用来证明一个假设是否成立,是获得可以用来否定与不否定这个假设的关键数据,或者事实。值得注意,临床试验获得的实验结果或数据用来形成进行统计学分析,最后得出结论,否定与不否定假设,而不是肯定某个假设。所以临床实验的设计必须获得足以进行统计学分析的数据,标本量,分析方法,主要观察数据等等。而NCT01096459新方案的主要设定结果:抓抓手术侧的皮肤能让膀胱产生15cm水柱以上的压力,而未手术侧的不能。 ( The primary aim of this study is reproducible bladder contraction of 15cm/H20 or more with scratching the cutaneous dermatome and no significant contraction with stimulation of the non-operated side )这是对肖氏手术进行效果评价的指标。同时标出术后6至24个月之后,手术是安全的。 . 在对主要设定结果的设计中,可能protocol没有达到同行的要求。要理解,这个临床试验是case series研究,而不是case-control 研究,做这样的试验,没有对照,压力大大啊!而且是一个Open Label,没有双盲,研究者自己评估自己的手术是否有显著性意义,你说这个压力大不大!!美国在1950年代的时候做过类似的临床试验,那就是脊髓灰质炎灭活疫苗(Salk疫苗),全民接种,几乎没有对照,可是那试验是罗斯福总统私募基金做的,跟NIH毫无关系。NIH的回复其实很含糊,根本没有谈将来会不会再启动资助,完全有可能principle investigator修改研究方案,继续申请NIH基金。 第三:Safety,安全性。NIH的回答根本没有说安全性问题,而是肯定NIH没有说过肖氏手术是安全的。在美国开展临床试验,安全性可是大头,每一个试验都要在方案中提前预见安全性及任何可能的副作用。抽血的风险就是minimal risk,因为受试者会有疼痛,可能会形成血肿,可能会引起出血和感染,这只是抽血。而肖氏手术是invasive procedure,也就是有创性的手术,而且在一个手术被认定处于试验阶段的时候,受试者可能获得的利益没有,研究方案或者知情同意书中如果写受试者可能获益(改善膀胱功能),这样的方案通过不了IRB的审查。而在可预见的风险里面:疼痛、疤痕、麻醉意外、出血、感染、神经瘤、神经损伤等等,这些都是major risk。尽管风险中没有死亡,但是死亡不是唯一的风险,一个病人如果试验之后一个月,走路不小心骨折,这也是不良反应。而且肖氏手术的受试者以儿童为主,这其中还牵涉到“易伤害人群,vulnerable population”,其伦理要求只会更加严格。肖氏手术的在现阶段的伦理价值之下,在权衡风险和利益比重之下,benefit/risk ratio,肖氏手术的美国临床试验之路依然充满崎岖与艰辛。 NIH的答复给肖氏手术最好的出路, more animal studies areneeded to demonstrate that the created reflex arc improves voiding. Ifthose are positive, then more mechanistic studies in animals would be needed toexplain how the arc functions, 更多的动物试验、更深入的机制研究。肖氏手术一定是中国医学史上重要的一笔,不管是美丽的,还是丑陋的。 ref: 美国NIH回答撤销肖氏手术临床试验的有关问题 http://blog.sciencenet.cn/blog-714-724252.html http://clinicaltrials.gov/ct2/show/NCT0109645
科学网网友 许培扬 上月曾就美国撤销肖氏手术临床试验的事情发过一篇博文,后来不知是否由于肖传国提出异议而撤掉了。 肖传国在科学网发博文“ NIH 扩大评估肖氏手术有效病种,继续资助肖氏手术研究” http://blog.sciencenet.cn/home.php?mod=spaceuid=385748do=blogid=713793 对为什么撤销临床研究做了解释。肖的博文还给出他称之为“真实情况”的一个网页链接 http://www.clinicaltrials.gov/ct2/results?term=rerouting 上述链接的网页上有两项临床试验与肖氏手术有关,一个名为 Lumbar to Sacral Ventral Nerve Re-Routing ( 临床试验识别号 NCT00378664) 仍在进行中,但不再招募患者。这是 2006 年开始的一项私人基金支持的研究,不是被撤销的临床试验。 被撤销的临床试验的识别号是 NCT01096459 ,名称为 Nerve Rerouting Treatment for Neurogenic Bladder in Spina Bifida 。这是美国政府出资支持的研究。由于对肖氏手术的争议,美国国家糖尿病暨消化系统及肾脏疾病研究所 (NationalInstitute of Diabetes, Digestive and Kidney Diseases, 简称 NIDDK) 在 2010 年底暂停了该临床试验,要求对治疗方法做进一步的评估。 《科学》曾对要求重新评估做过报道:“来自中国的疑问给美国的临床试验带来麻烦”。 http://blog.sciencenet.cn/blog-714-380535.html 评估的结果导致了肖氏手术的临床试验上月被撤销。临床试验信息网站上给出的理由是 Protocol as presented by investigator was not approved by the NIDDK. 研究者提出的治疗方案未被 NIDDK 批准。 NIDDK 上周五回答了我就撤销临床试验提出的几个问题, 全部登载并翻译如下。 Q: Why was the proposal not approved? Was it safety concerns? What are the specific reasons? 问 : 为什么提出的治疗方案没有获得批准?是出于安全考虑吗?有哪些具体原因? A: The Institute discussed the proposed study protocol (a prospective case-series study of surgical nerve rerouting in children with spina bifida) and received advice from experts at other institutes at NIH and an externalexpert panel. The experts thought that the proposed trial and its primary outcome measures were inadequately designed. After an extensive review, NIDDK decided not to fund the prospective case-series study of surgical nerve rerouting in children with spina bifida. 答 : 研究所讨论了该项目提出的治疗方案 ( 一项对脊柱裂儿童进行外科神经改道的前瞻性病例分析研究 ) ,收到了来自 NIH 内部其他研究所的专家、以及外部专家组的建议。专家们认为,研究提出的治疗方案、以及衡量其主要结果的指标设计不足。经过广泛评估之后, NIDDK 决定不支持这项对脊柱裂儿童进行外科神经改道的前瞻性病例分析研究。 Q: Now that the trial will not advance, will the trial continue to get funded? 问 : 现在临床试验不再进行,对这项临床研究的资助是否仍继续下去? A: NIH is no longer funding the trial, “Nerve ReroutingTreatment for Neurogenic Bladder in Spina Bifida”. 答 : NIH 不再资助这项名为“ Nerve ReroutingTreatment for Neurogenic Bladder in Spina Bifida ”的临床试验。 Q: What will the money already received be used for? 问 : 项目已经得到的经费将如何使用? A: The investigators at Beaumont Hospital in Royal Oak, Michigan, are able to use NIDDK funds to study the longer-term effects of nerve rerouting in children who had the nerve rerouting operation performed in a previous study that was not supported by the U.S. government. 答 : 密歇根州 RoyalOak 市的威廉 · 博蒙特医院的研究者能把来自 NIDDK 的经费用于研究儿童神经改道的长期效果上,这些儿童在此前一项不是由美国政府资助的研究中已经接受了神经改道的手术。 Q: What is NIDDK’s perspective regarding the nerve re-routing procedure? 问 : NIDDK 怎么看神经改道手术? A: In addition to research on the long-term effects that the investigators will examine (described above ) , more animal studies are needed to demonstrate that the created reflex arc improves voiding. If those are positive, then more mechanistic studies in animals would be needed to explain how the arc functions. 答 : 除了上面提到对其长期效果的研究之外,还需要更多的动物研究来证实建立起来的反射弧改善了排尿。如果动物实验结果是肯定的,仍需要在动物身上进行更多的机制性研究,以解释反射弧如何工作的问题。 Q: Is the following statement, posted on Dr. Xiao’s blog, true or false? The NIH has affirmed the safety of the procedure and considered the number of surgeries carried out so far statistically sufficient; therefore, the planned trial which will enroll 15 additional patients has been withdrawn. The study will continue as planned with further funding for the next two years. 问 : 肖传国博士在其博客上写道:“ NIH 已经认定肖氏手术安全”,而且“认定现有 SB 肖氏手术例数及结果已足够得出统计学结论,本研究不需再增加病例数;所以将原计划请 NIH 出资再作 15 例的部分撤销,改成现在的题目和内容继续进行、资助。”请问这否属实? A: No, NIH did not affirm the safety of the procedure or indicate that the number of surgeries is statistically sufficient. NIDDK feels the safety and efficacy of this experimental surgical intervention are unknown. 答 : 不属实。 NIH 没有认定过手术的安全性,或表示过已有的手术例数有统计学的意义。 NIDDK 认为用这个试验手术进行干预的安全性和效果尚属未知。
检索策略和检索结果: Found 56 studies with search of: 白血病 leukemia and 砒霜(三氧化二砷)arsenic trioxide 详细数据见: http://clinicaltrials.gov/ct2/results?term=leukemia+AND+arsenic+trioxidepg=1 Query Suggestions: The term, leukemia and arsenic trioxide, must be relaxed or trimmed to find documents Relax term to: leukemia AND arsenic trioxide leukemia AND arsenic trioxide Trim term to: arsenic trioxide arsenic trioxide Trim term to: leukemia leukemia Recognized Terms and Synonyms: leukemia and arsenic trioxide: 0studies arsenic trioxide: 97studies arsenic sesquioxide arsenous acid anhydride arsenous anhydride arsenous oxide as2o3 diarsenic trioxide naonobin trisenox white arsenic leukemia: 4274studies leucocythaemias leukemias trioxide: 111studies arsenic: 128studies as element Related Terms: Click on a related term to refine your search. This will narrow your search by adding an additional search term. If your search becomes too narrow (finds too few studies), broaden it by removing search terms with the Refine Search page. A acute promyelocytic leukemia aida regimen all trans retinoic acid apl apl-r arsenic sesquioxide arsenic trioxide arsenic(iii) oxide arsenous acid anhydride as2o3 aspergum aspirtab atra B buffex E easprin entaprin entercote F fab m3 G genacote genprin L leukemias acute promyelocytic leukemia M m3/m3 magnaprin minitabs N nrx 195183 P pml postremission therapy R rara T tamibarotene tretinoin trisenox V vesanoid Z zorprin 全部临床试验报告: study_fields.txt
全球临床试验研究项目计量分析报告 ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. ClinicalTrials.gov临床试验网 currently lists 137,539 studies 项目数量with locations in all 50 states and in 182 countries国家数量. ClinicalTrials.gov receives more than 95 million page views per month and 60,000 unique visitors daily 日访问量(as of February 2012). Contents Locations of Registered Studies Locations of Recruiting Studies Map of Studies Registered on ClinicalTrials.gov Types of Registered Studies Number of Registered Studies over Time Number of Registered Studies with Posted Results over Time Locations of Registered Studies This chart shows the distribution of locations for all studies registered on ClinicalTrials.gov. Total N = 137,539 studies (Data as of December 18, 2012) Legend for Recruiting Study location pie chart image Pie Color Location Non-U.S. Only (43%) U.S. Only (41%) Not Specified (9%) Both U.S. Non-U.S. (6%) Distribution of locations for all studies registered on ClinicalTrials.gov" Location Number of Registered Studies and Percentage of Total Non-U.S. Only 58,698 (43%) U.S. Only 56,983 (41%) Not Specified* 12,983 (9%) Both U.S. Non-U.S. 8,875 (6%) Total 137,539 * Not Specified: The location of the study was not provided by the Sponsor. (Data as of December 18, 2012) Locations of Recruiting Studies This chart shows the distribution of locations for recruiting studies registered on ClinicalTrials.gov. Total N = 29,400 studies (Data as of December 18, 2012) Legend for Recruiting Study location pie chart image Pie Color Location Non-U.S. Only (49%) U.S. Only (45%) Both U.S. Non-U.S. (6%) Distribution of locations for recruiting studies registered on ClinicalTrials.gov Location Number of Recruiting Studies and Percentage of Total Non-U.S. Only 14,378 (49%) U.S. Only 13,118 (45%) Both U.S. Non-U.S. 1,904 (6%) Total 29,400 (Data as of December 18, 2012) To Top Map of Studies Registered on ClinicalTrials.gov See Studies on a Map for an interactive map of studies that are registered on ClinicalTrials.gov. To Top Types of Registered Studies This table shows the number and types of studies that are registered and for which results are available on ClinicalTrials.gov. (Data as of December 18, 2012) Study and intervention types with number of registered studies and with number of studies having posted results Study and Intervention Type Number of Registered Studies and Percentage of Total Number of Studies With Posted Results and Percentage of Total*** Total 137,539 7,679 Interventional 111,598 (81%) 7,159 (93%) Type of Intervention* Drug or biologic 75,606 6,133 Behavioral, other 26,611 750 Surgical procedure 12,574 293 Device** 9,490 639 Observational 25,315 (18%) 520 (6%) Expanded Access 186 N/A * A study may include more than one type of intervention (that is, a single study may be counted more than once). Because of this, the sum of counts by type of intervention do not equal the total number of interventional studies. ** A total of 440 applicable device clinical trials were submitted as "delayed posting" under the Food and Drug Administration Amendments Act of 2007 (FDAAA). That is, the Responsible Party has indicated that such a trial includes a device not previously approved or cleared by the U.S. FDA for any use. These are not included in the count of trials with at least one device. *** Results are only required to be submitted for certain trials. For example, results submission is generally not required for trials of drugs, biologics, or devices that have not been approved by FDA; observational studies; and trials completed before 2008. N/A = not applicable To Top Number of Registered Studies Over Time The graph below shows the total number of studies registered on ClinicalTrials.gov since 2000, based on the First Received Date. The first version of ClinicalTrials.gov was made available to the public on February 29, 2000. (Data as of December 18, 2012) Key ICMJE: Indicates when the International Committee of Medical Journal Editors began requiring trial registration as a condition of publication under the Uniform Requirements for Manuscripts Submitted to Biomedical Journals (URM) (September 2005). FDAAA: Indicates when the expanded registration requirements of FDAAA began and were implemented on ClinicalTrials.gov (December 2007). Years with numbers of registered studies Year Total Number of Registered Studies 2000 5,647 2001 7,000 2002 8,598 2003 10,279 2004 12,081 2005 25,041 2006 35,984 2007 49,399 2008 66,448 2009 83,636 2010 101,356 2011 119,482 (Data as of December 18, 2012) To Top Number of Registered Studies With Posted Results Over Time The graph below shows the number of registered studies with results posted on ClinicalTrials.gov, based on the Results First Received Date. ClinicalTrials.gov launched its results database in September 2008, at which time sponsors or investigators were allowed to begin submitting results for their registered studies. The results database was developed to accommodate results submission requirements outlined in FDAAA. (Data as of December 18, 2012) Years with numbers of registered studies having posted results Year Total Number of Registered Studies With Posted Results 2009 1,795 2010 3,459 2011 5,742 (Data as of December 18, 2012) This page last reviewed in August 2012 To Top 数据来源: http://clinicaltrials.gov/ct2/resources/trends See Studies on Map Map of All Studies in ClinicalTrials.gov Click on the map below to show a more detailed map (when available) or search for studies (when map not available). Region Name Number of Studies World 137539 Africa 3094 Central America 1827 East Asia 11668 Japan 2555 Europe 36903 Middle East 5582 North America 72205 Canada 10420 Mexico 1772 United States 65858 North Asia 2530 Pacifica 3834 South America 4650 South Asia 2472 Southeast Asia 2763 Hints: Click on a link to show a map of that region Click on a link to search for studies in that region. Use the back button to return to this list and try another region. Studies with no locations are not included in the counts or on the map. Studies with multiple locations are included in each region containing locations. See Studies on Map Map of All Studies in ClinicalTrials.gov Click on the map below to show a more detailed map (when available) or search for studies (when map not available). Region Name Number of Studies World 137539 East Asia 11668 China 3434 Hong Kong 771 Korea, Republic of 3902 Mongolia 6 Taiwan 2836 Hints: Click on a link to show a map of that region Click on a link to search for studies in that region. Use the back button to return to this list and try another region. Studies with no locations are not included in the counts or on the map. Studies with multiple locations are included in each region containing locations. 数据来源: http://clinicaltrials.gov/ct2/search/map?map=ES
维生素A 缺乏症的临床试验研究有21项(见下面的详细信息),唐研究员做的研究课题很普通,研究目的、研究方法和研究结果没有什么特别的。从PUBMED数据库中检索到维生素A缺乏症的临床试验文献有269篇(见下面的信息分析结果)。 转基因大米人体试验(唐广文的论文和临床试验报告) 唐广文的论文昭示儿童转基因大米实验背景 (建议大家读一读) http://blog.sciencenet.cn/home.php?mod=spaceuid=280034do=blogid=609726 论文昭示儿童转基因大米实验背景 http://www.ebiotrade.com/newsf/2012-9/201295142556143.htm 该课题组的具体研究分工: 1 唐广文教授设计实验,实施田间试验,监督数据分析,并编写了手稿; 2 胡余明研究员协调和监督研究;荫士安进行研究和收集样品; 3 王茵研究员审核研究设计和监督研究;Gerard E Dallal监督和实施统计分析; 4 Michael A Grusak生产内源性标记的水稻和菠菜; 5 Robert M Russell设计了这项研究,并担任研究医生。 研究是在中国湖南省的一所小学开展的,对象是年龄在6-8岁的健康小学生(有着正常的生化检测结果;如下),无寄生虫感染,或用阿苯达唑(葛兰素史克)治疗后证实无感染。大部分居民是本地农村的劳动人民,收入中等。48%的研究对象在治疗(无副作用)后被招募,在研究餐开始前的一个月参与研究。因此,研究时的所有研究对象经过实验室检测,均无寄生虫感染。当地每年对所有儿童进行健康评估和生物医学检测(白细胞计数、血红蛋白计数、血小板计数、红细胞计数和红细胞压积)。生物通 www.ebiotrade.com 为了评估研究的样本大小,我们使用了过去美国的菠菜或β-胡萝卜素胶囊研究(26,27)和中国的深绿色蔬菜研究(28)进行power计算。我们发现,为了确定两个类别的差异——边缘和正常维生素A摄入,我们需要每个类别30个的样本大小,为了评估各种来源的食物机制的效果(菠菜、黄金大米和β-胡萝卜素胶囊),我们需要每组10个。考虑到对象退出研究的可能性,我们提议边缘维生素A组有36个对象,正常维生素A组有36个对象,而菠菜组、大米组和β-胡萝卜素胶囊组每组有12个。 在筛查的总共112个对象中,72个参加了研究,并从68个对象中采集了足够的血清样本(每个对象在1-5个时间点之间变化),便于HPLC和气相色谱-质谱分析。对象的人体测量是在全中国这一年龄组所报告的数据范围内(29)。 研究招募过程和程序经过了美国机构审查委员会-塔尔茨医学中心和中国浙江省医学科学院伦理审查委员会的批准。所有家长和学生都同意参加本研究。 致谢: 我们感谢所有中国湖南参与这一研究的志愿者,以及主厨、护士和中国长沙的湖南省疾病预防与控制中心相关人员。 作者分工如下: Guangwen Tang负责实验设计,实施田间实验,监督数据分析并且撰写论文 Yuming Hu:协调并监督这项研究 Shi-an Yin:进行研究并收集样本 Yin Wang:评估实验设计并进行监督 Gerard E Dallal:监督并进行统计学分析 Michael A 黄金大米usak:生产带标记的大米和菠菜 Robert M Russell:设计实验并作为本研究中的医师 所有的作者都看过论文手稿,没有作者与该研究存在利益冲突。 http://blog.sciencenet.cn/blog-280034-609409.html 2012年最新的这篇题为“β-Carotene in Golden Rice is as good as β-carotene in oil at providing vitamin A to children”的文章一经过公布,就引起了众多关注,文章主要是对儿童进行试验,试验分三组,一共有68个年龄6-8岁的儿童,在35天时间里,三组儿童分别摄入这种试验大米,菠菜,和β-胡萝卜素的胶囊。结论是在提供给儿童维生素A的方面,GR大米中β-胡萝卜素与胶囊中的一样有效,并且优于菠菜。 关于“Golden Rice”(转基因黄金大米,GR)的报道近期掀起了轩然大波,目前最新进展是相关文章(β-Carotene in Golden Rice is as good as β-carotene in oil at providing vitamin A to children)中第二,第三和第四作者(均为中方作者)都否认了曾参与这项研究,称“之前既没有听说与该篇论文相关的任何信息,也没有看过论文的内容,更不知道为何自己的名字会出现在论文的作者之中”,事件进展扑朔迷离,在进一步调查结果出来之前,让我们来看看相关发表的论文。 http://www.ebiotrade.com/newsf/2012-9/201295115024123.htm 安德莉亚·格罗斯曼在声明中说 ,“这项试验的目的是测试黄金大米作为在发展中国家一个非常严重健康问题(维生素A缺乏)的部分解决方案的可行性。” 维生素A 缺乏症是因体内缺乏维生素A而引起的以眼和皮肤病变为主的全身性疾病,多见于1~4岁小儿;最早的症状是暗适应差,眼结合膜及角膜干燥,以后发展为角膜软化且有皮肤干燥和毛囊角化,故又称夜盲症、干眼病、角膜软化症。 Vitamin A Equivalence of Plant Carotenoids in Children This study has been completed. Study NCT00680212 Information provided by Tufts University First Received on May 16, 2008. Last Updated on February 17, 2009 History of Changes Related Studies can be found by searching for the Conditions, Interventions, and Sponsors found in this study: Conditions listed in this trial Vitamin A Deficiency Additional conditions recognized in this trial Night Blindness More general conditions related to this trial Avitaminosis Deficiency Diseases Eye Diseases Malnutrition Nutrition Disorders Vision Disorders Interventions listed in this trial dietary carotenoids Additional drug interventions recognized in this trial Beta Carotene Carotenoids Retinol palmitate Vitamin A Vitamins More general drug interventions related to this trial Anticarcinogenic Agents Antineoplastic Agents Antioxidants Growth Substances Micronutrients Molecular Mechanisms of Pharmacological Action Pharmacologic Actions Physiological Effects of Drugs Protective Agents Therapeutic Uses Sponsors listed in this trial Tufts University http://clinicaltrials.gov/ct2/show/related/NCT00680212 Found 21 studies with search of: "Vitamin A Deficiency" Hide studies that are not seeking new volunteers. Hide studies with unknown recruitment status. Display Options Rank Status Study 1 Completed Treatment of Congenital Stationary Night Blindness With an Alga Containing High Dose of Beta Carotene Condition: NightBlindness Intervention: DietarySupplement:algaDunaliellabardawil 2 Completed Efficacy of Vitamin A in Fortified Extruded Rice in School Children Condition: VitaminADeficiency Intervention: Dietary Supplement: Triple fortified extruded tice (Fe, Zn and vitamin A) 3 Recruiting Efficacy of Newborn Vitamin A Supplementation in Improving Immune Function Condition: VitaminADeficiency Interventions: DietarySupplement:retinylpalmitate; DietarySupplement:Placebo 4 Recruiting Efficacy of Yellow Cassava to Improve Vitamin A Status of Kenyan School Children Condition: VitaminADeficiency Interventions: Other:Yellowcassava; Other:Whitecassava 5 Completed An Efficacy Trial of Iron, Zinc and Vitamin A Fortified Rice in Children in Satun, Thailand Conditions: VitaminADeficiency; IronDeficiency; ZincDeficiency Intervention: DietarySupplement:fortifiedextrudedrice 6 Completed Impact of Maternal Supplementation With Dual Megadose of Vitamin A Conditions: Hypovitaminosis; VitaminADeficiency Interventions: DietarySupplement:vitaminA; DietarySupplement:Placebo 7 Active, not recruiting Vitamin A Bioavailability in Lactating Women With Marginal Vitamin A Status Condition: VitaminADeficiency Interventions: Other:0mgretinolactivityequivalents; Other:12mgofBC; Other:6mgofCX; Other:1.0mgRAE 8 Completed Vitamin A to Reduce HIV in Vaginal Secretions and Prevent Viral Transmission Conditions: HIVInfections; VitaminADeficiency; HIVSeronegativity Intervention: Drug:VitaminA 9 Completed Vitamin A Equivalence of Plant Carotenoids in Children Condition: VitaminADeficiency Interventions: DietarySupplement:dietarycarotenoids; Dietary Supplement: spinach, rice, and synthetic beta-carotene 10 Unknown † Single-dose Postpartum Vitamin A Supplementation of Mothers and Neonates Conditions: VitaminADeficiency; HIV Intervention: Drug:VitaminA(retinylpalmitate) 11 Completed Retinol Equivalence of Plant Carotenoids in Children Condition: VitaminADeficiency Intervention: 12 Completed Has Results Impact of Maternal Vitamin A or Beta-Carotene Supplementation on Maternal and Infant Mortality in Bangladesh Conditions: VitaminADeficiency; MaternalMortality; InfantMortality Intervention: Dietary Supplement: Vitamin A or Beta-Carotene Supplements 13 Completed Trial of the Impact of Vitamin A on Maternal Mortality Conditions: VitaminADeficiency; MaternalMortality; MaternalMorbidity Intervention: DietarySupplement:VitaminA 14 Completed Impact of Consumption of Orange-fleshed Sweet Potatoes on the Vitamin A Status of Bangladeshi Women of Reproductive Age Condition: VitaminADeficiency Intervention: Behavioral: Consumption of orange-fleshed sweet potatoes 15 Terminated Community Trial of Newborn Vitamin A Supplementation to Reduce Infant Mortality in Rural Bangladesh Conditions: InfantMortality; VitaminADeficiency Intervention: Drug:vitaminAsupplementation(50,000IU) 16 Recruiting Effect of Vitamin A Supplementation on Immune Responses in Human Neonates Condition: VitaminADeficiency Intervention: Dietary Supplement: Vitamin A (retinyl palmitate). 17 Completed Dietary Vitamin A Requirement in Chinese Children and the New Technology of Dietary Assessment Condition: VitaminADeficiency Intervention: 18 Completed Vitamin A Absorption From Cassava in Women Condition: VitaminADeficiency Interventions: Other: Beta-Carotene bio-fortified cassava porridge without oil; Other: Beta-Carotene bio-fortified cassava porridge with oil; Other: White cassava porridge with retinyl palmitate reference dose 19 Completed Newborn Vitamin A (VA) Supplementation Pilot Project, Pakistan Condition: VitaminADeficiency Interventions: DietarySupplement:VitaminA; DietarySupplement:Placebo 20 Active, not recruiting Nutributter Programming to Prevent Undernutrition: an Evaluation Condition: Undernutrition Interventions: DietarySupplement:NutributterProgram; Other:IntegratedPackage Found 21 studies with search of: "Vitamin A Deficiency" Hide studies that are not seeking new volunteers. Hide studies with unknown recruitment status. Display Options Rank Status Study 21 Completed A Study on Immunological Effect of Vitamin A and Zinc in a Placebo Controlled 4 Cell Trial Conditions: Immunity; Diarrhea; RespiratoryTractInfections Intervention: Drug:Zinc,orVitaminAorboth 详细信息见 http://clinicaltrials.gov/ct2/results?cond=%22Vitamin+A+Deficiency%22pg=1 Found 21 studies with search of: "Vitamin A Deficiency" No Query Suggestions Recognized Terms and Synonyms: Vitamin A Deficiency: 21studies deficiencies disease vitamins deficiencies vitamins deficiency of vitamin a hypovitaminosis a keratomalacia night blindness retinol deficiency vitamin a deficiencies vitamin deficiency disease xerotic keratitis Related Terms: Click on a related term to refine your search. This will narrow your search by adding an additional search term. If your search becomes too narrow (finds too few studies), broaden it by removing search terms with the Refine Search page. B beta-carotene C carotenes cassava N night blindness O of vitamin a deficiency P palmitates retinol palmitate provitamin a R retinol retinol activity equivalent retinol equivalent retinol palmitate retinyl palmitate S spinach V vitamin a deficiencies of vitamin a deficiency vitamin a intake 检索策略: 1 "vitamin a deficiency维生素A缺乏" OR "vitamin a deficiency" 检索结果 5619篇 2 ("vitamin a deficiency" OR "vitamin a deficiency" ) AND Clinical Trial临床试验 检索结果269篇 信息分析平台: http://arrowsmith.psych.uic.edu/cgi-bin/arrowsmith_uic/AnneOTate_summarize.cgi?ID=31269t=fw=ch 科研论文信息分析结果: Important words 重要词汇 词频Frq 1 vitamin 4661 2 retinol 1250 3 xerophthalmia 287 4 bitot 116 5 hypovitaminosis 216 6 deficiency 3097 7 retinyl 255 8 provitamin 113 9 keratomalacia 60 10 vad 252 11 micronutrient 244 12 mrdr 24 13 deficient 1259 14 carotene 353 15 avitaminosis 89 16 xerophthalmic 24 17 retinoic 514 18 a-deficient 41 19 blindness 368 20 supplementation 664 Topics 研究主题分布 Frq 1 Vitamin A Deficiency 4525 2 Vitamin A 2643 3 Liver 718 4 Diet 454 5 Xerophthalmia 370 6 Tretinoin 350 7 Nutritional Status 342 8 Body Weight 306 9 Carotenoids 306 10 Prevalence 289 11 Nutrition Disorders 258 12 India 253 13 Rats, Inbred Strains 252 14 Dietary Supplements 235 15 Developing Countries 217 16 Retinol-Binding Proteins 215 17 Risk Factors 209 18 Cross-Sectional Studies 195 19 Night Blindness 190 20 Iron 179 Author 作者分布 frq 1 Sommer A 86 2 West KP 58 3 Ross AC 53 4 Semba RD 52 5 Wolf G 48 6 West CE 44 7 Muhilal 36 8 Olson JA 36 9 De Luca LM 35 10 Biesalski HK 32 11 Reddy V 32 12 Amedee-Manesme O 30 13 DeLuca HF 29 14 Underwood BA 29 15 Bloem MW 27 16 Tanumihardjo SA 26 17 Katz J 25 18 Russell RM 25 19 Zile MH 24 20 Chytil F 23 Affiliations 研究机构分布 frq 1 USA 420 2 Baltimore 128 3 India 115 4 France 95 5 Department of Biochemistry 93 6 Department of Ophthalmology 92 7 Department of Pediatrics 91 8 Japan 66 9 Boston 61 10 London 53 11 Department of Nutrition 52 12 The Netherlands 52 13 Canada 48 14 UK 46 15 New York 45 16 MD 21205 44 17 Brazil 38 18 Department of Human Nutrition 38 19 Department of International Health 38 20 Department of Medicine 37 Journals 期刊分布 frq 1 J Nutr 280 2 Am J Clin Nutr 230 3 Int J Vitam Nutr Res 117 4 Nutr Rev 91 5 Lancet 85 6 Br J Nutr 76 7 Eur J Clin Nutr 68 8 Biochem J 63 9 Indian Pediatr 61 10 Arch Latinoam Nutr 50 11 Biochim Biophys Acta 49 12 Food Nutr Bull 49 13 Vopr Pitan 49 14 Ukr Biokhim Zh 47 15 Invest Ophthalmol Vis Sci 42 16 J Trop Pediatr 42 17 Public Health Nutr 42 18 Br J Ophthalmol 40 19 Indian J Pediatr 40 20 Proc Soc Exp Biol Med 35 Years年代分布 1 2012 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 2 2011 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 3 2010 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 4 2009 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 5 2008 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 6 2007 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 7 2006 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 8 2005 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 9 2004 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 10 2003 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 11 2002 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 12 2001 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 13 2000 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 14 1999 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 15 1998 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 16 1997 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 17 1996 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 18 1995 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 19 1994 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx 20 1993 xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx Topics 文献聚类 Frq Most recent articles 347 1 Liver 718 2 Diet 454 3 Xerophthalmia 370 4 Tretinoin 350 5 Nutritional Status 342 6 Carotenoids 306 7 Body Weight 306 8 Prevalence 289 9 Nutrition Disorders 258 10 India 253 11 Rats, Inbred Strains 252 12 Developing Countries 217 13 Retinol-Binding Proteins 215 14 Epithelium 178 15 Vitamin E Deficiency 168 Miscellaneous 2133 参考文献 • 评论:谁隐瞒了“儿童试验 转基因 大米”真相 • 原论文全文: 转基因 大米儿童实验(一) • 利用儿童实验的 转基因 大米论文全文(三) • 湖南农业厅:从未批准 转基因 大米试验 • 转基因 大米试验部分涉事家长称不知是试验 • 利用儿童实验的 转基因 大米论文全文(四) • 利用儿童实验的 转基因 大米论文全文(二) • 一论文利用中国儿童做 转基因 试验引争议 • 转基因 研究难以避免人体试验 • 转基因 大米人体试验中美当事人说法不一致 • 国家动物 转基因 技术研究中心落户内蒙古大学 • 转基因 大米试验发生了什么 • 转基因 大米不是毒,但是是风险,以及无罪推定 • 小学生 转基因 试验陷“罗生门” • 评论: 转基因 大米试验需要实质性回应 • 美大学承认用中国儿童做 转基因 大米试验 • 制备 转基因 小鼠过程中的若干技巧 • 湖南衡阳称无学生涉及 转基因 人体试验 • 转基因 水稻的基因逃逸“不可避免但缓慢” • 两头 转基因 “克隆牛”在京出生喝进口奶粉 • 科学家研制出首例 转基因 芒草 • 农业 转基因 管理各国迥异 • 转基因 玉米:有望在中国大放光芒 • 全世界78%的 转基因 作物生产自美洲大陆 • 害虫突变可能会危及 转基因 作物的功效 • 美国新一代 转基因 大豆悄然上市FDA为其开绿灯 • 转基因 细菌阻止蚊子传播疟疾 • 盖茨基金会千万美元资助英国 转基因 农业研究 • 美国民众网络请愿拒绝“ 转基因 蚊子” • 用 转基因 蚊子解决登革热等问题 • “消费者无需担忧有“准生证”的 转基因 食品” • 袁隆平称不应该全否定 转基因 • 中国 转基因 主粮安全问题是是非非十年 • 美国科学家培育出世界上第一批 转基因 婴儿 • 疾控专家:有“准生证”的 转基因 食品可放心吃 • 元勋子女斗法 转基因 • 中国 转基因 稻违规扩散 • 转基因 大豆油成北京学校食堂主力
我建议:在没有取得充分证据前,科学网的博主和读者不要急于下结论,先仔细看看唐广文研究员的原始论文和临床试验报告。 ----- 许博主 美国塔夫茨大学回应:正在调查 本报驻美国记者吴成良 记者9月1日向唐广文(音)博士发去电子邮件,就“黄金大米”试验疑云请其作出澄清。记者在9月2日收到了塔夫茨大学健康科学中心公共关系助理主任安德莉亚·格罗斯曼发来的电邮,就该事件做了声明,同时附上了“唐广文和她的同事2012年8月1日和2009年先后发表在《美国临床营养学期刊》(AJCN)网站上的两篇相关论文”。 安德莉亚·格罗斯曼在声明中说,“这项试验的目的是测试黄金大米作为在发展中国家一个非常严重健康问题(维生素A缺乏)的部分解决方案的可行性。” 格罗斯曼在声明中说:“这项研究聚焦一小组健康的中国儿童,年龄在6至8岁。中国儿童血液中维生素A含量低是普遍现象。试验前征得了所有儿童家长的同意。所有参与试验的儿童都接受了一定形式的维生素A。” 但这份声明较为含糊。声明仅称参与试验的儿童都接受了一定形式的维生素A,未说明试验的儿童是否吃了黄金大米。 美国当地时间9月4日上午10时左右,本报驻美国记者接到格罗斯曼亲自打来的电话。格罗斯曼称,正在召集相关部门人员开会了解此事,这需要一定时间,因为他们需要在联系有关方面和人员之后才能给出回应,而唐广文暂时不在办公室。最快可能要到当地时间4日下午才能给记者回复。 http://international.dbw.cn/system/2012/09/05/054211328_03.shtml 当事人否认转基因大米试验使用“黄金大米”唐广文被疑学术造假 http://env.people.com.cn/n/2012/0905/c1010-18925670.html 2012 American Society for Nutrition β-Carotene in Golden Rice is as good as β-carotene in oil at providing vitamin A to children 1 , 2 , 3 , 4 Guangwen Tang , Yuming Hu , Shi-an Yin , Yin Wang , Gerard E Dallal , Michael A Grusak , and Robert M Russell + Author Affiliations 1 From the Carotenoids Health Laboratory, USDA Human Nutrition Research Center on Aging at Tufts University, Boston, MA (GT, GED, and RMR); the Hunan Province Center for Disease Control and Prevention, Changsha, China (YH); Maternal Child Nutrition, National Institute for Nutrition and Food Safety, Beijing, China (SY); the Zhejiang Academy of Medical Sciences, Hangzhou, China (YW); the USDA-ARS Children's Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX (MAG); and the Office of Dietary Supplement s , NIH, Bethesda, MD (RMR). + Author Notes ↵ 2 Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the US Department of Agriculture, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. ↵ 3 This material is based on work supported by the US Department of Agriculture under Cooperative Agreements 581950-9-001, 58-6250-6-001, and 581950-7-707; by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases, NIH (NIDDK DK620021); and by a grant on spinach and pure labeled β-carotene from the National Technology Research and Development Program in the 11th Five Year Plan of China (2008BAI58B03). ↵ 4 Address correspondence to G Tang, Jean Mayer USDA, Human Nutrition Research Center on Aging, Tufts University, 711 Washington Street, Boston, MA 02111. E-mail: guangwen.tang@tufts.edu . Abstract Background: Golden Rice (GR) has been genetically engineered to be rich in β-carotene for use as a source of vitamin A. Objective: The objective was to compare the vitamin A value of β-carotene in GR and in spinach with that of pure β-carotene in oil when consumed by children. Design: Children ( n = 68; age 6–8 y) were randomly assigned to consume GR or spinach (both grown in a nutrient solution containing 23 atom% 2 H 2 O) or β-carotene in an oil capsule. The GR and spinach β-carotene were enriched with deuterium ( 2 H) with the highest abundance molecular mass (M) at M β-C + 2 H 10 . Retinyl acetate in an oil capsule was administered as a reference dose. Serum samples collected from subjects were analyzed by using gas chromatography electron-capture negative chemical ionization mass spectrometry for the enrichments of labeled retinol: M retinol +4 (from β-carotene in oil), M retinol +5 (from GR or spinach β-carotene), and M retinol +10 (from retinyl acetate). Results: Using the response to the dose of retinyl acetate (0.5 mg) as a reference, our results (with the use of AUC of molar enrichment at days 1, 3, 7, 14, and 21 after the labeled doses) showed that the conversions of pure β-carotene (0.5 mg), GR β-carotene (0.6 mg), and spinach β-carotene (1.4 mg) to retinol were 2.0, 2.3, and 7.5 to 1 by weight, respectively. Conclusions: The β-carotene in GR is as effective as pure β-carotene in oil and better than that in spinach at providing vitamin A to children. A bowl of ∼100 to 150 g cooked GR (50 g dry weight) can provide ∼60% of the Chinese Recommended Nutrient Intake of vitamin A for 6–8-y-old children. This trial was registered at www.clinicaltrials.gov as NCT00680212 . Received November 17, 2011. Accepted June 21, 2012. http://ajcn.nutrition.org/content/96/3/658.abstract 8月31日,有网文称,美国一专业网站刊登的论文透露,美国塔夫茨大学一科研机构2008年在湖南省一所小学进行过转基因大米(黄金大米)人体试验,该网文随即在国内外引发强烈关注。 记者了解到,8月1日,《美国临床营养学杂志》网站发表了一篇名为《黄金大米中的β-胡萝卜素与油胶囊中的β-胡萝卜素对儿童补充维生素A同样有效》的论文。 论文称,为了比较儿童摄入“黄金大米”、菠菜和β-胡萝卜素油胶囊对补充维生素A有何不同,美国塔夫茨大学、湖南疾病预防控制中心、中国疾控中心营养与食品安全所、浙江医学科学院等工作机构的研究人员2008年共同在湖南省的一所小学进行试验,针对的是6到8岁的健康的在校小学生。 论文同时称,研究所用材料——黄金大米和菠菜都是在美国生产、处理和蒸煮,然后冷藏运至中国实验所在地加热后供小学生食用。 论文称,所有的作者均审查了原稿。 该论文共有7名作者。据记者了解,论文第一作者唐广文(音)为美国塔夫茨大学研究员,论文第二作者胡余明为中国湖南省疾控中心工作人员,第三作者荫士安为中国疾控中心研究员,第四作者王茵为浙江医学科学院研究人员。其他3位作者为杰拉德·戴罗尔、米切尔·格鲁萨克、罗伯特·罗素。另据了解,所谓“黄金大米”是一种转基因大米,因呈黄色而得名。 http://international.dbw.cn/system/2012/09/05/054211328.shtml 人民日报:美国转基因大米实验食材来自湖南当地 发布时间: 2012-09-05 07:45来源: 人民日报 进入电子报 http://news.cnhubei.com/xw/gn/201209/t2213536.shtml 美国课题组织者承认给湖南小学生吃转基因大米 http://news.hf365.com/system/2012/09/05/011732039_01.shtml 美高校回应“黄金大米”事件:试验征得中方家长同意 http://news.cnnb.com.cn/system/2012/09/05/007446371.shtml Vitamin A Equivalence of Plant Carotenoids in Children This study has been completed. First Received on May 16, 2008. Last Updated on February 17, 2009 History of Changes Sponsor: Tufts University Information provided by: Tufts University ClinicalTrials.gov Identifier: NCT00680212 Purpose Our objectives will be to test the following hypotheses and to make the following determinations: (1) The absorption and bio-conversion of provitamin A carotenes taken by children are different between spinach, Golden Rice, and -C in oil capsules. (2) The absorption of provitamin A carotenes and their bioconversion to vitamin A are different in children with or without adequate vitamin A nutrition. (3) To define the vitamin A equivalence(s) of dietary spinach, Golden Rice, and a -C in oil dose by using an isotope reference method in children with or without adequate vitamin A nutrition and to compare those values with values derived from model based compartmental analysis. (4) To determine the number and time of blood samples needed for future studies in various field settings on the retinol equivalence of a large number of plant sources. Condition Intervention Phase Vitamin A Deficiency Dietary Supplement: dietary carotenoids Dietary Supplement: spinach, rice, and synthetic beta-carotene Phase 2 Study Type: Interventional Study Design: Allocation:Randomized EndpointClassification:Bio-equivalenceStudy InterventionModel:FactorialAssignment Masking:OpenLabel PrimaryPurpose:BasicScience Official Title: Phase 2 Study of VITAMIN A EQUIVALENCE OF PLANT CAROTENOIDS IN CHILDREN Resource links provided by NLM: Genetics Home Reference related topics: carbamoylphosphatesynthetaseIdeficiency MedlinePlus related topics: VitaminA Drug Information available for: Retinol VitaminApalmitate betacarotene U.S. FDA Resources Further study details as provided by Tufts University: Primary Outcome Measures: conversion efficiency of b-C to retinol Estimated Enrollment: 72 Study Start Date: July 2008 Study Completion Date: January 2009 Primary Completion Date: January 2009 (Final data collection date for primary outcome measure) Arms Assigned Interventions 1 dietary carotenoids Dietary Supplement: dietary carotenoids spinach containing 1 - 2 mg beta-carotene rice containing 0.5 mg beta-carotene synthetic beta-carotene 0.5 mg oil capsule Dietary Supplement: spinach, rice, and synthetic beta-carotene spinach containing 1 - 2 mg beta-carotene rice containing 0.5 mg beta-carotene synthetic beta-carotene, 0.5 mg oil dose Other Name: dietary carotenoids Detailed Description: This project is to determine the vitamin A value (equivalence) of dietary provitamin A carotenes from spinach, Golden Rice, and pure -carotene (-C) in oil. These experiments will be conducted in children (ages 6-8) with/without adequate (marginal deficiency) vitamin A nutrition. As plant provitamin A carotenoids are a major and safe vitamin A source for a vast population in the world, it is essential to determine the efficiency of provitamin A carotenoid (mainly -C) conversion to vitamin A. By introducing -C into rice endosperm, Golden Rice may directly benefit consumers by providing vitamin A nutrition. Our investigation uses hydroponically grown, deca-deuterium labeled spinach and Golden Rice, synthetic -C-d10 and a vitamin A isotope reference, C13 labeled retinyl acetate (13C10-RAc), to evaluate the bioavailability and the bioconversion of plant provitamin A carotenes to retinol as compared with -C in oil capsules in vivo. Seventy-two children each will take two meals, breakfast containing 13C10-RAc dose (0.5mg in 0.2g oil capsule) and lunch containing spinach containing 1 mg -C (along with white rice), or Golden Rice containing 0.5mg -C (along with light colored vegetables), or -C oil capsules containing 0.5 mg -C in 0.2g oil (along with white rice and light colored vegetables) on the first day of the study. Blood samples will be collected at 1 3, 7, 14, and 21 days after the study doses. The enrichment of labeled -C and labeled retinol in human circulation will be determined using advanced liquid chromatography / mass spectrometry and gas chromatography / mass spectrometry. Through the applications of these novel technologies, we will be able to determine the relative biological activities of endogenous carotenoids; that is, the vitamin A value of spinach, Golden Rice, and -C in oil capsules for children with/without vitamin A malnutrition. This study will be of importance in planning vitamin A deficiency prevention strategies and also will provide useful information regarding the potential efficacy of a bioengineered crop to provide vitamin A nutrition. Eligibility Ages Eligible for Study: 6 Years to 8 Years Genders Eligible for Study: Both Accepts Healthy Volunteers: Yes Criteria Inclusion Criteria: healthy children Exclusion Criteria: food allergy parasitic infection Contacts and Locations Please refer to this study by its ClinicalTrials.gov identifier: NCT00680212 Locations United States, Massachusetts USDA Human Nutrition Research Center on Aging, Tufts Uni. Boston, Massachusetts, United States, 02111 Sponsors and Collaborators Tufts University Investigators Principal Investigator: Guangwen Tang, Ph. D Tufts University More Information No publications provided Responsible Party: Guangwen Tang, Tufts University ClinicalTrials.gov Identifier: NCT00680212 History of Changes Other Study ID Numbers: IRB 8458, R01DK60021 Study First Received: May 16, 2008 Last Updated: February 17, 2009 Health Authority: United States: Institutional Review Board Keywords provided by Tufts University: dietary beta-carotene vitamin A status intrinsically labeled plant foods Additional relevant MeSH terms: Vitamin A Deficiency Night Blindness Avitaminosis Deficiency Diseases Malnutrition Nutrition Disorders Vision Disorders Eye Diseases Carotenoids Retinol palmitate Vitamin A Vitamins Beta Carotene Antioxidants Molecular Mechanisms of Pharmacological Action Pharmacologic Actions Protective Agents Physiological Effects of Drugs Micronutrients Growth Substances Anticarcinogenic Agents Antineoplastic Agents Therapeutic Uses ClinicalTrials.gov processed this record on September 03, 2012 http://clinicaltrials.gov/show/NCT00680212 《美国临床营养学期刊》 American Journal of Clinical Nutrition http://www.ajcn.org/ Country : United States Subject Area : Agricultural and Biological Sciences | Medicine Subject Category : Food Science , Medicine (miscellaneous) Publisher : American Society for Clinical Nutrition, Inc. . Publication type : Journals. ISSN : 00029165, 19383207 Coverage: 1952-2011 影响因子: 6.669 H Index: 186 Scope: The purpose of The American Journal of Clinical Nutrition (AJCN) is to publish original research studies relevant to human and The purpose of The American Journal of Clinical Nutrition (AJCN) is to publish original research studies relevant to human and clinical nutrition. Well-controlled clinical studies that describe scientific mechanisms, efficacy, and safety of dietary interventions in the context of disease prevention or a health benefit will be considered. Public health and epidemiologic studies relevant to human nutrition, and innovative investigations of nutritional questions that employ epigenetic, genomic, proteomic, and metabolomic approaches are encouraged. Solicited editorials,book reviews, solicited or unsolicited review articles, invited controversy position papers, and letters to the Editor that relate to prior AJCN articles are essential components of the AJCN. ( source ) Show full scope
二期临床是2009年五月发表(人数199人,剂量是两组:10mg, 20mg),三期临床的文章是6月发表的,都是四军大西京医院的赵医生(G. Zhao)主持。标题上说的是多中心的研究。但是总人数不到400,而且在接受治疗的病人中,中度和重度病人占比较低,因此样本量不是很大,对于脑中风这种病种,这样的病人数好像是少了点。从临床研究数据来看,GSRD还是有效果的。该研究没有比较性研究。总的来说,数据还是单薄了点。 该研究注明的资助单位是:Tai-He Biopharmaceutical Co. Ltd. (Guangzhou, P.R. China). 另:查了一些人参皂苷的资料,三七里面人参皂苷_Rd的含量很低,单纯从植物里面提取,成本不小的。现在还没有方法进行化学合成,据现有资料表明,现在比较好的方法是首先获得各种人参皂苷的亚型,比如Rb1,Rb2等,它们的侧链和Rd不一样,需要通过一些微生物表达的酶通过酶促反应,将其转化成Rd。化学药物里面,有些药物因为合成困难,难以产业化,人参皂苷-Rd不知道这方面具体如何,需要进一步研究。GSRD为何这么受关注,是因为它是特异的受体拮抗剂,可以阻断经受体型通道进入的钙电流(查文献应该是TRP受体),这种电流一般持续时间长,对细胞损伤很大。 三期临床数据 数据来源文献: X. Liu,et al, Ginsenoside-Rd improves outcome of acute ischaemic stroke – a randomized, double-blind, placebo-controlled, multicenter trial,European Journal of Neurology, 2012,19(6):855-863. 临床实验效果统计分析采用了两种方法,一是改良型Rankin指数(Modified Rankin Scale, mRs)和NIHSS。 mRs的统计结果如下: a为全样本分析,b为有效样本分析。 mRs系统采用分级来说明病人的临床症状。级别越小,说明病人的病情症状越轻,0是没有症状,1-2基本上可以独立照顾自己,3-4为中度,生活不能完全自理,需要协助,5是严重缺乏自理能力,需要全天候陪护。 按照全样本分析数据,与对照组(Placebo)相比,人参皂苷-Rd组(GSRD)在分级上出现了一定程度的改善。0-2级比例,安慰组为64%,干预组为80%,有一定程度提升;3-4级比例,安慰组33%,干预组14%,显著下降;5-6级,安慰组5%,干预组2%,显著下降。有效样本的分布比例类似。 如果从病人症状的迁徙率上看,干预组病人症状从重症向中度,或者从中度向轻度迁徙的比率是非常明显的。该试验共有病人386人,其中对照组96人,干预组290人。该试验中由于中度和中度患者的人数较小,因此还不能很好的表明GSRD的临床效果。另外没有现有临床用药的对比,因此不能评价GSRD的相对效果是否有提升,不能说明Me Better. 其它分析方法: NIHSS(NIH中风评分,小于10为轻度)的评分,处理组有77%的患者在10分一下,安慰组为78%。腔隙性梗死(Lacunar infarct)没有明显差别。下图不大看得懂,作者说NIHSS分数在处理组有明显改善,P值小于0.01.置信区间0.95。 其它图表: 原文链接: http://onlinelibrary.wiley.com/doi/10.1111/j.1468-1331.2011.03634.x/full 临床指标简介: The Modified Rankin Scale (mRS) The scale runs from 0-6, running from perfect health without symptoms to death . 0 - No symptoms. 1 - No significant disability. Able to carry out all usual activities, despite some symptoms. 2 - Slight disability. Able to look after own affairs without assistance, but unable to carry out all previous activities. 3 - Moderate disability. Requires some help, but able to walk unassisted. 4 - Moderately severe disability. Unable to attend to own bodily needs without assistance, and unable to walk unassisted. 5 - Severe disability. Requires constant nursing care and attention, bedridden, incontinent. 6 – Dead OCSP分类法 The Oxford Community Stroke Project classification (OCSP, also known as the Bamford or Oxford classification) relies primarily on the initial symptoms. Based on the extent of the symptoms, the stroke episode is classified as total anterior circulation infarct (TACI), partial anterior circulation infarct (PACI), lacunar infarct (LACI) or posterior circulation infarct (POCI). These four entities predict the extent of the stroke, the area of the brain affected, the underlying cause, and the prognosis.
出版空间以及编辑关注度的竞争异常激烈。将原稿投送给杂志编辑,附上一封信“原稿请见附件”是远远不够的。投稿信是你与拟投杂志直接交流的机会。除了写明你的研究与众不同外,还应直接向总编辑说明为什么你的发现很重要及其应该在此杂志上发表的理由。 投 稿信应含有几个重要内容。具体内容可通过www.liwenbianji.cn/coverletter 链接下载。大家可根据批注中的建议起草你自己的投稿信,选择提出的句子类型替代括号中的句子。投稿信的格式几乎适用于所有投稿;当然,某些类别的论文需要 加入额外的内容。例如,关于临床试验数据的存储信息通常需要附上一份临床试验报告,提供你的序列数据进入公共数据库的信息。 查阅目标杂志 的《稿约》是每篇稿件的既定程序,其中很可能含有投稿信必须写入的内容。另外一个信息来源是杂志的投稿网页。尽管以下列出的内容以及关于“Edanz投稿 信模版”中描述的内容不一定完全都是这些目标杂志所要求的,但所有这些都是投稿信中必不可少的,因为这样做可引起编辑对你的关注。以下方法适用于投稿信的 撰写: • 一些杂志根据其刊出文章领域的不同进行编辑分工,你可以根据不同的领域,有时也可根据编辑的专业背景选择最合适的编辑。直接称呼收信编辑,如:“Dear Dr. Smith”。如果不能找到合适的编辑,可将投稿信写给总编辑。 • 信的开头应写出文章题目,希望文章在杂志的哪一个栏目或作为哪一个文章类别发表,以及投稿杂志的名称。 • 之后简单叙述研究背景与理论基础,说明研究目的以及开展的工作。然后简单描述研究成果。 • 接下来的段落很重要。你需要向研究界解释你的发现的意义,特别是对杂志读者的意义。如果你不能解释为什么该杂志读者会对你的发现感兴趣,你需要选择另一家 更合适的刊物,因为编辑只将他们认为会引起读者兴趣的文章送同行评议。研究一下你准备投稿杂志的“目标与刊出范围”会对你有帮助。 • 投稿信的最后一段应包含杂志所要求的声明或说明。这些通常包括关于利益冲突、基金资助与资助来源的声明,以及所有作者已阅读过并同意文章的内容以及未一稿多投的声明。每个作者的作者资格确认也是需要的。 • 最后,留下详细的通讯方式以及礼貌的结束语。 示例: 英文原文 The cover letter: your sales pitch Competition for publication space and for editors’ attention is now very high, and it is no longer sufficient to send a manuscript to a journal editor along with a letter saying little more than “please find my manuscript attached”. The cover letter is your opportunity to directly address the editor of your target journal. It can be used to set your study apart from others and directly explain to the editor why your findings are important and why they should be published in their journal. There are a number of important components of a cover letter, all of which should be included. These components are described in detail in Edanz Cover Letter Template, which is shown on the following page and can be downloaded from: www.liwenbianji.cn/coverletter. This template can be used to develop your own cover letters by following the suggestions in the comments and replacing the bracketed sentences with the types of sentences explained. The format of this letter is applicable for most if not all submissions, although additional sections may be required for some types of paper; for example, information about deposition of clinical trial data would most likely need to accompany a report of a clinical trial, and information about the deposition of sequence data into public databases would possibly need to be provided where such data has been obtained. As always, the target journal’s instructions to authors should be consulted; these will most likely outline the information that absolutely must be included in the cover letter. Another source of this information is the journal’s submission webpages. Although not all of the components listed below and described in the cover letter template will be described as required on the target journal’s webpages, all should be included in your letter, because to do so will increase your chances of grabbing the editor’s attention. The following principles apply to cover letter development: • Some journals have different editors for the different areas of research the journal covers and you can choose the most appropriate one based on area and occasionally also editor profiles. Address your letter personally to the appropriate editor, e.g., “Dear Dr. Smith”. If one cannot be readily identified, address your letter to the editor-in-chief. • Begin by providing the title of your manuscript, the section/publication type you would like to see it published as, and the name of the journal you are submitting it to. • You then need to provide a very brief background and rationale for your study, explaining why you did what you did. This can be followed by a brief description of the results. • The following paragraph is very important. You will need to explain the significance of your findings to the research community, and specifically to the readers of your target journal. If you find it difficult to explain why the readers of that journal would be interested in your findings, then you may need to select a more appropriate journal. Editors will only send papers to review that they think will be of interest to their readers. Studying the ‘aims and scope’ of your chosen journal might help with this. • The last paragraph of the letter should contain any statements or declarations required by the target journal. These usually include declarations of any conflicts of interest, grant support or other sources of funding, a statement that all authors have read and approved the manuscript and a statement that the same manuscript has not been submitted elsewhere. Confirmation of each author’s qualification for authorship may also be required. • Finally, include details for correspondence and a polite farewell. Example: Dr Daniel McGowan 分子神经学博士 理文编辑学术总监
Hi, Off label and open label are different. Off label : Getting the drug outside the trial, if you have been on it for a diffeent indication and responded , the drug is made available to you. Open label : Trial withone or two drugs where the content of the drug is known both to the patient and the doctor. No blind drug but in a trial. Drug may be available commercially on prescription as well. Dr Sharat C Misra MD,DM,FACG Re:What Here we go again: Open label , open trial : all know the drug Single Blind trial : Patient doesnt know the drug, Doc knows the drug he is giving Double blind trial : Neither the Doc nor the patient know the drug, they are labelled A and B and the codes are in a sealed envelope , decoded only after the trial, usually by the company suppying the drug or the trial co-ordinator. Dr Sharat C Misra MD,DM,FACG http://www.hbvhbv.com/forum/thread-64342-1-1.html
氢气生物学研究目前进展迅速,虽然有大量的动物实验证明对许多疾病具有治疗作用,但如果没有严格的随机对照临床试验的证据,则无法获得临床治疗特别是国家医药管理局等的最终许可,也就是说无法获得官方的正式批准用于临床的使用。开展严格对照的临床研究是氢气医学发展最重要的任务和手段。也是将来深入研究氢气生物学效应最重要的环境保障和研究目的。 但国际上在临床试验方面进展并不快,到目前为止,临床试验的报道基本都来自日本,这里是从世界卫生组织临床试验注册的信息中检索到的临床试验注册信息也说明这个问题,这些信息显示出日本在神经系统损伤治疗方面的关注度比较大,例如 6 项注册试验中有 3 项属于神经系统损伤治疗效果的研究,分别是中风、巴金森病和中度认知障碍的研究。比较有意思的是,最早报道氢气生物学效应的日本医科大学没有注册临床试验的信息,是他们没有信心,还是没有获得研究经费的资助。因为他们曾经获得来自商业公司的赞助,并成立氢气医学中心。从这一点上看,似乎没有这些问题。但内情不清楚。 第一项:氢气水治疗巴金森病 第二项:氢气水对正常人的抗氧化效果评价 第三项 氢气生理盐水注射对脑缺血的治疗效果评价 第四项 氢气水对中度认知障碍治疗效果的研究 第五项 氢气水治疗间质性膀胱炎 第六项 氢气水对糖尿病的治疗效果评价 所有信息可从世界卫生组织的临床试验注册网上免费检索,更详细的信息可从 http://apps.who.int/trialsearch/AdvSearch.aspx 检索。 建议检索词为 : hydrogen 。 Recruitment status 选择 ALL 。否则无法获得全面的信息。 详细信息 第一项:氢气水治疗巴金森病 2012 年 3 月 14 日注册的用“氢气水治疗巴金森病”开始实验 2010 年 1 月 1 日顺天堂大学附属医院神经外科,联系人 Asako Yoritaka 。日本学者曾经报道使用氢气水治疗巴金森病的动物实验效果,发表论文 3 篇。全部使用自由饮用氢气水。治疗设计 The subjects should make 1000 ml of molecular hydrogen water which contains 1.6 ppm dissolved hydrogen by Aquerable, and consume for 48 weeks . Placebo water which is not contained molecular hydrogen water made from pseudo-machine. The subjects consume for48 weeks. 第二项:氢气水对正常人的抗氧化效果评价 Studies on in vivo effects of drinking a water product dissolving hydrogen gas as an in vitro antioxidant additive 杏林大学 Atsushi Hiraoka 自 2009 年 5 月 1 日开始的针对健康人的一项研究,排除肝脏肾脏功能异常和月经期女性。 Ingestion of 500ml per day of hydrogen gas-dissolving water for 1 week.Ingestion of 500ml per day of control water without hydrogen gas for 1 week.. 观察指标 the levels of serum LPO and urine 8-OHdG in subjects immediately before and after 1-week drinking period for 500ml per day of tap water with or without dissolved hydrogen gas at 0.34mg/l and 1-week before and after the drinking per day. 第三项 氢气生理盐水注射对脑缺血的治疗效果评价 日本国防医科大学神经外科 Hiroshi Nawashiro 于 2011/06/01 开始的 Molecular hydrogen for ischemic stroke 。选择诊断后症状发生 24 小时内脑缺血患者 Patients were eligible for enrollment if they were 18 years or older and had a clinical diagnosis of acute ischemic stroke within 24 hours of symptom onset. They had to score at least 6 on the National Institutes of Health Stroke Scale (NIHSS) with at least 2 points for limb weakness. All patients received appropriate routine stroke care as per local treatment practices, including alteplase for eligible patients presenting 3 hours from onset; patients receiving alteplase had to commence the study drug before the alteplase infusion.Exclusion criteria: Patients with acute ischemic stroke beyond 24 hours of symptom onset. 治疗方法为静脉点滴注射氢气生理盐水。效果评价 modified Rankin scale (mRS) (days 7, 30, and 90), the NIHSS (days 7 and 90), and the Barthel Index (days 7, 30, and 90) Safety Assessments Vital signs were recorded at enrollment and at specified times throughout the infusion and follow-up periods. Routine laboratory data and ECGs were performed at the time of enrollment, at 24 and 72 hours, and on day 7 and were analyzed centrally (ECGs at day 7 were performed only if abnormal at 72 hours). To assess any effect of hydrogen on hemorrhagic transformation after alteplase administration, brain imaging was repeated after 72 hours in patients who were receiving concomitant treatment with alteplase. Symptomatic hemorrhagic transformation was defined as an increase in the NIHSS score of at least 4 points within 36 hours, plus evidence of any blood on neuroimaging after treatment with alteplase. Patients meeting criteria for progressive stroke (NIHSS increase of 4 points within 72 hours) or new stroke in the first week were also reimaged. 第四项 氢气水对中度认知障碍治疗效果的研究 筑波医科大学临床医学研究所神经精神学系 Takashi Asada2009/07/01 开始的 A randomized trial to assess the effects of hydrogen-ride dissolution water for the patients with mild cognitive impairment (MCI). 中度认知障碍的研究。 招募对象: Inclusion criteria: 1) Participants of the Tone project. 2) Being able to give a written informed consent to the participation in the present study. 3) Having diagnosis of the mild cognitive impairment. 4) Being able to observe the following requirement: good compliance with the water; participation in the scheduled examinations for assessment; keeping a log-diary recording the consumption of the water. 5) Having a modified Hachinski Ischemic score of 4 or less. 6) Having the 15-item Geriatric Depression Scale score of 6 or less. Exclusion criteria: 1) Meeting DSM-IV TR criteria for dementing illnesses. 2) Having serious or unstable illnesses. 3) Having a history within past 5 years of serious infectious disease affecting the brain and/or malignant diseases. 4) Having a history of alcohol or drug abuse or dependence (on DSM-IV TR) within the past 5 years. 5) Receiving any types of anti-Alzheimer drugs. 6) Recent (within 4 weeks) initiation of medications that affect the central nervous system.Age minimum: 67years-old.Age maximum: Not applicable Gender: Male and Female 研究内容: mild cognitive impairment 治疗手段 The patients of hydrogen group will be intervened with 500ml hydrogen dissolution water every-day for 2 years. The patients of placebo group will be intervened with 500ml ordinary water every-day for 2 years. 效果评价 Score in Japanese version of ADAS-Cog and Mini Mental State Examination. Scores in Japanese version of ADCS-ADL, MRI and SPCET imaging, and Geriatric Depression Scale. 第五项 氢气水治疗间质性膀胱炎 Koushinkai Hospital 的 Comprehensive Support Project for Clinical Research Office 于 2008/07/01 开始的 A randomized trial to asses the effects of hydrogen-rich dissolution water in patients with interstitial cystitis 。至少现在没有氢气在间质性膀胱炎方面的研究报道,无论是动物实验还是临床报道。 研究对象标准: Inclusion criteria: 1) Patients who are able to give written informed consent 2) Patients who has the characteristic finding under hydraulic distension of the bladder in interstitial cystitis by cystoscope 3) It has taken more than 12 weeks after patients took the hydraulic distension of the bladder, and symptom of patients are in stable. 4) More than 7 marks in total of Interstitial Cystitis Score in registration 5) More than 4 marks in Q4 (degree of bladder pain) in Interstitial Symptom Score 6) Age is over 20 years and less than 80 years 7) Patients are able to do the following things in this trial; getting good compliance with intaking investigating food and coming to hospital, and writing the diary and the questionnaire accurately by themselvesExclusion criteria: 1) More than 200ml of an average voided volume at a time before the registration 2) Patients with active infection of urinary tract 3) Patients with bacterial cystitis within 12 weeks before registration 4) Patients with vaginosis 5) Patients with calculus of lower urinary tract or urethral diverticulum 6) Patients with nephrosis syndrome 7) Patients with active genital herpes 8) Patients who have operated the surgery in pelvis or its circumference and it has not taken more than 24 hours after the surgery 9) Patients with cerebrospinal disease 10) Patients with the follow disease or suspected disease; neurogenic bladder, cystitis radiation, tuberculous cystitis, cystitis with BCG, drug associated cystitis 11) Start, stop, or change of the dose of the following drugs within 4 weeks after the registration; (a) Antiphlogistic analgetic (b) Antidepressant (c) Anticholinergic drug (d) Antihistamine drug (e) Ataractic drug (f) Drug treatment for frequent urination and acraturesis (g) Steroid drug 12) Start or stop new bladder training or diet therapy within 4 weeks befor registration 13) Patients who has received bladder instillation therapy, electrical stimulation therapy, or acupuncture and moxibustion within 12 weeks before registration 14) Patients with serious hepatic or kidney damage 15) Patients with serious heart disease 16) Patients with malignant tumors which effect their general status or survival time 17) Patients with the history of serious drug-induced adverse effect 18) Patients who are in pregnancy, while breast-feeding, or have possibilities of them, or desire pregnancy in test period 19) Patients who have taken part in the her clinical research within 12 weeks 20) Patients who have taken part in the her clinical research within 12 weeks 21) Patients who are inadequate, which their physicians assessed itAge minimum: 20years-old Age maximum: 80years-oldGender: Male and Female 治疗方法 The patients will be intervened with hydrogen dissolution water group (hydrogen group) 200ml every three times in a day in 2 months (56days) . After that, the patients in hydrogen dissolution water group will be transferred to the additional intervention term after the end of intervention.And after that, the patients will be randomized to withdrawal terms for more 1 month with hydrogen dissolution water or with placebo water. The proportion of the patients who has been assessed "success" at the end of the intervention Secondary Outcome(s) 1) Changes of the Symptom Score in Interstitial Cystitis Symptom Index(ICSI) 2) Changes of the Problem Score in Interstitial Cystitis Problem Index(ICPI) 3) An Average frequency of urination per day 4) An Average voided volume at a time 5) Degree of urge to urinate; PUF symptom score 6) Degree of bladder pain 7) Impression by patients with GRA (Global Response Assessment) 8) Urine Test; 8-OHdG in urine 9) Adverse Events (we cannot deny the association between the food and the event) 第六项 氢气水对糖尿病的治疗效果评价 研究京都大学医学院 Comprehensive Support Project for Clinical Research Office 于 21/08/2008 开始的 A Randomized trial to assess the effects of hydrogen-rich dissolution water for patients with impaired glucose tolerance or impaired fasting glucose 。该研究已经发表论文。 研究对象标准: Inclusion criteria: 1) Patients who are abele to give written informed consent 2) FBS is over 100mg/dl and under 126mg/dl in registration 3) Age is over 20 years and less than 80 years 4) Type of practice: outpatient department 5) Patients are able to do the following things in this trial - getting good compliance with consuming investigational food and coming to hospital, and writing the diary and the questionnaire accurately by themselvesExclusion criteria: 1) Patients who have receive drug treatment for diabetes 2) Patients with the diseases which have possibility with impaired glucose tolerance 3) Patients with serious liver or kidney damage 4) Patients with serious heart disease or cerebrovascular disorders 5) Patients with serious disease in pancreas or blood disease 6) Patients with malignant tumors which effect their general status or survival time 7) Patients who are in pregnancy, while breast-feeding, have possibilities of them, or desire pregnancy in test period 8) Patients with alcohol abuse 9) Patients who have taken part in the her clinical research within 12 weeks 10) Patients who have taken part in the her clinical research within 12 weeks 11) Patients who are inadequate, which their physicians assessed it Age minimum: 20years-oldAge maximum: 70years-oldGender: Male and Female 研究内容 Impaired glucose tolerance or impaired fasting glucose 治疗手段 The patients will be intervened with hydrogen dissolution water group (hydrogen group) 200ml every three times in a day in 3 months (84days) . The patients will be intervened with normal water group (placebo group) 200ml every three times in a day in 3 months (84days) 效果评价方法 1)75gOGTT(glucose);0 minute (in the fasting state),30,60,90 minutes later after loading2)Delta AUC(0-120min);The difference of the area under the plasma glucose concentration before and after administration
出版空间以及编辑关注度的竞争异常激烈。将原稿投送给杂志编辑,附上一封信“原稿请见附件”是远远不够的。投稿信是你与拟投杂志直接交流的机会。除了写明你的研究与众不同外,还应直接向总编辑说明为什么你的发现很重要及其应该在此杂志上发表的理由。 投 稿信应含有几个重要内容。具体内容可通过www.liwenbianji.cn/coverletter 链接下载。大家可根据批注中的建议起草你自己的投稿信,选择提出的句子类型替代括号中的句子。投稿信的格式几乎适用于所有投稿;当然,某些类别的论文需要 加入额外的内容。例如,关于临床试验数据的存储信息通常需要附上一份临床试验报告,提供你的序列数据进入公共数据库的信息。 查阅目标杂志 的《稿约》是每篇稿件的既定程序,其中很可能含有投稿信必须写入的内容。另外一个信息来源是杂志的投稿网页。尽管以下列出的内容以及关于“Edanz投稿 信模版”中描述的内容不一定完全都是这些目标杂志所要求的,但所有这些都是投稿信中必不可少的,因为这样做可引起编辑对你的关注。以下方法适用于投稿信的 撰写: • 一些杂志根据其刊出文章领域的不同进行编辑分工,你可以根据不同的领域,有时也可根据编辑的专业背景选择最合适的编辑。直接称呼收信编辑,如:“Dear Dr. Smith”。如果不能找到合适的编辑,可将投稿信写给总编辑。 • 信的开头应写出文章题目,希望文章在杂志的哪一个栏目或作为哪一个文章类别发表,以及投稿杂志的名称。 • 之后简单叙述研究背景与理论基础,说明研究目的以及开展的工作。然后简单描述研究成果。 • 接下来的段落很重要。你需要向研究界解释你的发现的意义,特别是对杂志读者的意义。如果你不能解释为什么该杂志读者会对你的发现感兴趣,你需要选择另一家 更合适的刊物,因为编辑只将他们认为会引起读者兴趣的文章送同行评议。研究一下你准备投稿杂志的“目标与刊出范围”会对你有帮助。 • 投稿信的最后一段应包含杂志所要求的声明或说明。这些通常包括关于利益冲突、基金资助与资助来源的声明,以及所有作者已阅读过并同意文章的内容以及未一稿多投的声明。每个作者的作者资格确认也是需要的。 • 最后,留下详细的通讯方式以及礼貌的结束语。 示例: 英文原文 The cover letter: your sales pitch Competition for publication space and for editors’ attention is now very high, and it is no longer sufficient to send a manuscript to a journal editor along with a letter saying little more than “please find my manuscript attached”. The cover letter is your opportunity to directly address the editor of your target journal. It can be used to set your study apart from others and directly explain to the editor why your findings are important and why they should be published in their journal. There are a number of important components of a cover letter, all of which should be included. These components are described in detail in Edanz Cover Letter Template, which is shown on the following page and can be downloaded from: www.liwenbianji.cn/coverletter. This template can be used to develop your own cover letters by following the suggestions in the comments and replacing the bracketed sentences with the types of sentences explained. The format of this letter is applicable for most if not all submissions, although additional sections may be required for some types of paper; for example, information about deposition of clinical trial data would most likely need to accompany a report of a clinical trial, and information about the deposition of sequence data into public databases would possibly need to be provided where such data has been obtained. As always, the target journal’s instructions to authors should be consulted; these will most likely outline the information that absolutely must be included in the cover letter. Another source of this information is the journal’s submission webpages. Although not all of the components listed below and described in the cover letter template will be described as required on the target journal’s webpages, all should be included in your letter, because to do so will increase your chances of grabbing the editor’s attention. The following principles apply to cover letter development: • Some journals have different editors for the different areas of research the journal covers and you can choose the most appropriate one based on area and occasionally also editor profiles. Address your letter personally to the appropriate editor, e.g., “Dear Dr. Smith”. If one cannot be readily identified, address your letter to the editor-in-chief. • Begin by providing the title of your manuscript, the section/publication type you would like to see it published as, and the name of the journal you are submitting it to. • You then need to provide a very brief background and rationale for your study, explaining why you did what you did. This can be followed by a brief description of the results. • The following paragraph is very important. You will need to explain the significance of your findings to the research community, and specifically to the readers of your target journal. If you find it difficult to explain why the readers of that journal would be interested in your findings, then you may need to select a more appropriate journal. Editors will only send papers to review that they think will be of interest to their readers. Studying the ‘aims and scope’ of your chosen journal might help with this. • The last paragraph of the letter should contain any statements or declarations required by the target journal. These usually include declarations of any conflicts of interest, grant support or other sources of funding, a statement that all authors have read and approved the manuscript and a statement that the same manuscript has not been submitted elsewhere. Confirmation of each author’s qualification for authorship may also be required. • Finally, include details for correspondence and a polite farewell. Example: Dr Daniel McGowan 分子神经学博士 理文编辑学术总监
分享一下2012年5月16日Biotronik公司在其网站上发布他们对镁合金药物洗脱支架的12个月临床(欧洲多中心,6个月和12个月2组共46个患者)随访结果,与去年发布的6个月结果相比,Twelve-month results from the BIOSOLVE-I study with the DRug Eluting Absorbable Metal Scaffold (DREAMS) demonstrate safety and confirm vessel vasomotion.这对做可降解金属的同仁们会是个鼓励。 pdf下载在这里 BIOTRONIK_PR%20BIOSOLVE-I_EuroPCR_EN.pdf http://www.biotronik.de/wps/wcm/connect/int_web/biotronik/newsroom/press_releases?p=http://www.biotronik.de/wps/wcm/connect/int_web/biotronik/newsroom/press_releases/press_release_biosolve_ipw=770pt = At one year, DREAMS demonstrated a low 7.0% rate of target lesion failure with no death or scaffold thrombosis. The target lesion revascularization rate was 4.7%. The analysis showed an in-scaffold late lumen loss of 0.52 mm, which was further reduced from the 6-month results. Vasoreactivity at 12 months did not significantly change from the 6-month results, suggesting that the natural physiology of the vessels was already restored after 6 months of healing and could be sustained. “The reduction of the 12-month late lumen loss is likely due to plaque regression and late expansive remodeling. At 6 months, we have already seen that vasomotion is restored, so the new results verify that the vessels are naturally adapting to accommodate flow,” said Professor Haude. “Additionally, we could further validate that the return of natural vessel angulation that we saw at 6 months was maintained also at 12 months. These results suggest a real restoration of the vessel’s natural architecture.” DREAMS is made of a proprietary magnesium alloy coated with a matrix of a bioabsorbable polymer and paclitaxel to inhibit neointimal cell proliferation within the first few months after scaffold implantation. Mechanical properties of the magnesium alloy allow deployment of the device to be comparable to conventional metallic stents. “The particularly positive feature of the magnesium scaffold is its nice conformability to the vessel wall. A few months after DREAMS implantation, the artery is able to regain its original shape and physiology. This is the main benefit of the magnesium scaffold over the more rigid polymer-based platforms,” Professor Haude added. “These positive one-year results confirm that we are working in the right direction. BIOTRONIK continues to invest in our DREAMS program as we believe that bringing vascular restoration therapy to patients without changing the way physicians implant a scaffold is important to the adoption of this technology,” stated Alain Aimonetti, Vice President of Sales and Marketing at BIOTRONIK Vascular Intervention. “An improvement of late lumen loss between 6 and 12 months shows us that the values become more predictable over time. It makes sense, especially for the bioabsorbable scaffolds, to focus on long-term outcomes and not so much on short-term results,” Aimonetti concluded. “We see huge potential for DREAMS since it combines deployment and postdilatation properties and long-term outcomes comparable to DES with the additional benefits of vascular restoration therapy. About the BIOSOLVE-I Clinical Trial The BIOSOLVE-I first-in-man study is a prospective, multicenter, nonrandomized, European, first-in-man clinical trial evaluating the safety and efficacy of DREAMS. Forty-six patients were enrolled in two cohorts that evaluated the primary endpoint of target lesion failure (TLF) at 6 months for cohort 1 and at 12 months for cohort 2. About the DRug Eluting Absorbable Metal Scaffold (DREAMS) DREAMS is part of a revolutionary new treatment option for patients with coronary artery disease . In contrast to existing permanent stents, this device is based on a slow-absorbing, high-strength magnesium backbone. The vessel is not caged as it would be with a permanent implant, thus the artery is able to resume its natural physiology. This novel treatment concept opens a new area of vascular restoration therapy. Properties of the proprietary magnesium alloy allow a straightforward deployment of the device, comparable to conventional metallic stents. BIOTRONIK’s DREAMS is coated with antiproliferative paclitaxel to inhibit excess tissue growth during the healing process. The company has announced next-generation platforms under development using the BIOlute coating from the Orsiro hybrid drug-eluting stent featuring sirolimus as the active compound. About BIOTRONIK SE Co. KG As one of the world’s leading cardiovascular medical device companies, with several million implanted devices, BIOTRONIK is represented in over 100 countries with its global workforce of more than 5,600 employees. Known for having its fingers on the pulse of the medical community, BIOTRONIK assesses the challenges physicians face and provides the best solutions for all phases of patient care, ranging from diagnosis and treatment to patient management. Quality, innovation and clinical excellence define BIOTRONIK and its growing success—and deliver confidence and peace of mind to physicians and their patients worldwide.
出版空间以及编辑关注度的竞争异常激烈。将原稿投送给杂志编辑,附上一封信“原稿请见附件”是远远不够的。投稿信是你与拟投杂志直接交流的机会。除了写明你的研究与众不同外,还应直接向总编辑说明为什么你的发现很重要及其应该在此杂志上发表的理由。 投稿信应含有几个重要内容。具体内容可通过www.liwenbianji.cn/coverletter 链接下载。大家可根据批注中的建议起草你自己的投稿信,选择提出的句子类型替代括号中的句子。投稿信的格式几乎适用于所有投稿;当然,某些类别的论文需要加入额外的内容。例如,关于临床试验数据的存储信息通常需要附上一份临床试验报告,提供你的序列数据进入公共数据库的信息。 查阅目标杂志的《稿约》是每篇稿件的既定程序,其中很可能含有投稿信必须写入的内容。另外一个信息来源是杂志的投稿网页。尽管以下列出的内容以及关于“Edanz投稿信模版”中描述的内容不一定完全都是这些目标杂志所要求的,但所有这些都是投稿信中必不可少的,因为这样做可引起编辑对你的关注。以下方法适用于投稿信的撰写: • 一些杂志根据其刊出文章领域的不同进行编辑分工,你可以根据不同的领域,有时也可根据编辑的专业背景选择最合适的编辑。直接称呼收信编辑,如:“Dear Dr. Smith”。如果不能找到合适的编辑,可将投稿信写给总编辑。 • 信的开头应写出文章题目,希望文章在杂志的哪一个栏目或作为哪一个文章类别发表,以及投稿杂志的名称。 • 之后简单叙述研究背景与理论基础,说明研究目的以及开展的工作。然后简单描述研究成果。 • 接下来的段落很重要。你需要向研究界解释你的发现的意义,特别是对杂志读者的意义。如果你不能解释为什么该杂志读者会对你的发现感兴趣,你需要选择另一家更合适的刊物,因为编辑只将他们认为会引起读者兴趣的文章送同行评议。研究一下你准备投稿杂志的“目标与刊出范围”会对你有帮助。 • 投稿信的最后一段应包含杂志所要求的声明或说明。这些通常包括关于利益冲突、基金资助与资助来源的声明,以及所有作者已阅读过并同意文章的内容以及未一稿多投的声明。每个作者的作者资格确认也是需要的。 • 最后,留下详细的通讯方式以及礼貌的结束语。 示例: 英文原文 The cover letter: your sales pitch Competition for publication space and for editors’ attention is now very high, and it is no longer sufficient to send a manuscript to a journal editor along with a letter saying little more than “please find my manuscript attached”. The cover letter is your opportunity to directly address the editor of your target journal. It can be used to set your study apart from others and directly explain to the editor why your findings are important and why they should be published in their journal. There are a number of important components of a cover letter, all of which should be included. These components are described in detail in Edanz Cover Letter Template, which is shown on the following page and can be downloaded from: www.liwenbianji.cn/coverletter. This template can be used to develop your own cover letters by following the suggestions in the comments and replacing the bracketed sentences with the types of sentences explained. The format of this letter is applicable for most if not all submissions, although additional sections may be required for some types of paper; for example, information about deposition of clinical trial data would most likely need to accompany a report of a clinical trial, and information about the deposition of sequence data into public databases would possibly need to be provided where such data has been obtained. As always, the target journal’s instructions to authors should be consulted; these will most likely outline the information that absolutely must be included in the cover letter. Another source of this information is the journal’s submission webpages. Although not all of the components listed below and described in the cover letter template will be described as required on the target journal’s webpages, all should be included in your letter, because to do so will increase your chances of grabbing the editor’s attention. The following principles apply to cover letter development: • Some journals have different editors for the different areas of research the journal covers and you can choose the most appropriate one based on area and occasionally also editor profiles. Address your letter personally to the appropriate editor, e.g., “Dear Dr. Smith”. If one cannot be readily identified, address your letter to the editor-in-chief. • Begin by providing the title of your manuscript, the section/publication type you would like to see it published as, and the name of the journal you are submitting it to. • You then need to provide a very brief background and rationale for your study, explaining why you did what you did. This can be followed by a brief description of the results. • The following paragraph is very important. You will need to explain the significance of your findings to the research community, and specifically to the readers of your target journal. If you find it difficult to explain why the readers of that journal would be interested in your findings, then you may need to select a more appropriate journal. Editors will only send papers to review that they think will be of interest to their readers. Studying the ‘aims and scope’ of your chosen journal might help with this. • The last paragraph of the letter should contain any statements or declarations required by the target journal. These usually include declarations of any conflicts of interest, grant support or other sources of funding, a statement that all authors have read and approved the manuscript and a statement that the same manuscript has not been submitted elsewhere. Confirmation of each author’s qualification for authorship may also be required. • Finally, include details for correspondence and a polite farewell. Example: Dr Daniel McGowan 分子神经学博士 理文编辑学术总监
本研究的亮点:采用随机、双盲、安慰剂对照,观察了氢气水对患者线粒体和炎症肌病(肌肉营养不良)的治疗效果。 分子氢气具有许多神奇的效应,超过 30 多种动物疾病模型中发现氢气具有治疗氧化应激相关疾病的作用。更重要的是,在临床研究中,人们发现氢气对人类二型糖尿病、血液透析、代谢综合征、肝癌放射治疗的损伤、脑干中风具有显著治疗效果。氢气治疗疾病的细胞学机制被认为是通过清除羟基自由基、过氧亚硝酸根这样具有强大氧化作用的自由基(不影响温和的过氧化氢等),而且氢气也可以影响细胞内信号系统。但是目前人们对氢气生物效应的分子细节仍不清楚(这里确实是一个非常令人值得期待的研究方向)。氢气是一个具有生物安全性的分子,人体内不停产生,到目前几乎没有任何明显不良作用被发现。因此氢气相关药物可能成为一类令人神往的临床应用药物。本文是第一个氢气医学领域的双盲随机对照研究。 先开展的开放性试验(试探性研究)中让进行性肌肉营养不良( 进行性肌萎缩 ) 患者每天饮 1 升富氢气水,连续 12 周。其中, 4 例患者为 多发性肌炎 - 皮肌炎(似乎是自身免疫性疾病), 5 例患者为线粒体病。每四周检测 18 类血清学指标和尿的人 8- 异前列腺素 F2a ( 8-isoprostane 是可以说明氧化损伤程度的指标)。在随后的随机、双盲、安慰剂试验中,有 10 例 DM( 肌病 ) 和 12 例 线粒体病患者,给患者连续 8 周(怎么没有用 12 周) 0.5 升富氢水(为什么不是 1 升)或普通水(安慰剂),每 4 周检测 18 类血清指标。 研究结果:在该开放性研究(患者和医生了解治疗情况的研究)中,没有发现临床症状的加重或减轻。但是,发现氢气在肌肉病患者中乳糖 / 丙酮酸比例,餐后血糖,血清 MMP3 和血清甘油三脂具有显著影响。在双盲试验中,也没有发现明显的临床效果,但 DM 患者的乳糖 / 丙酮酸比例,和 MM 患者的血清 MMP3 仍明显改变,尽管没有统计学差异。没有在糖尿病患者中发现任何不良效应。 结论:氢气水 MM 患者的对线粒体功能和 PM/DM 患者的炎症反应过程具有治疗效果,但双盲试验中效果较差的原因可能是剂量比较小和观察时间比较短(为什么不增加剂量,延长观察时间?)。但也说明氢气的效果存在一个最低有效剂量(意思是说不能喝太少,至少要 1 升,有广告嫌疑),而且有剂量效应(更确认效果的可靠性)。 Research Open-label trial and randomized, double-blind, placebo-controlled, crossover trial of hydrogen-enriched water for mitochondrial and inflammatory myopathies Mikako Ito , Tohru Ibi , Ko Sahashi , Masatoshi Ichihara , Masafumi Ito and Kinji Ohno For all author emails, please log on . Medical Gas Research 2011, 1 :24doi:10.1186/2045-9912-1-24 Published: 3 October 2011 Abstract (provisional) Background Molecular hydrogen has prominent effects on more than 30 animal models especially of oxidative stress-mediated diseases and inflammatory diseases. In addition, hydrogen effects on humans have been reported in diabetes mellitus type 2, hemodialysis, metabolic syndrome, radiotherapy for liver cancer, and brain stem infarction. Hydrogen effects are ascribed to specific radical-scavenging activities that eliminate hydroxyl radical and peroxynitrite, and also to signal-modulating activities, but the detailed molecular mechanisms still remain elusive. Hydrogen is a safe molecule that is largely produced by intestinal bacteria in rodents and humans, and no adverse effects have been documented. Methods We performed open-label trial of drinking 1.0 liter per day of hydrogen-enriched water for 12 weeks in five patients with progressive muscular dystrophy (PMD), four patients with polymyositis/dermatomyositis (PM/DM), and five patients with mitochondrial myopathies (MM), and measured 18 serum parameters as well as urinary 8-isoprostane every 4 weeks. We next conducted randomized, double-blind, placebo-controlled, crossover trial of 0.5 liter per day of hydrogen-enriched water or placebo water for 8 weeks in 10 patients with DM and 12 patients with MM, and measured 18 serum parameters every 4 weeks. Results In the open-label trial, no objective improvement or worsening of clinical symptoms was observed. We, however, observed significant effects in lactate-to-pyruvate ratios in PMD and MM, fasting blood glucose in PMD, serum matrix metalloproteinase-3 (MMP3) in PM/DM, and serum triglycerides in PM/DM. In the double-blind trial, no objective clinical effects were observed, but a significant improvement was detected in lactate in MM. Lactate-to-pyruvate ratios in MM and MMP3 in DM also exhibited favorable responses but without statistical significance. No adverse effect was observed in either trial except for hypoglycemic episodes in an insulin-treated MELAS patient, which subsided by reducing the insulin dose. Conclusions Hydrogen-enriched water improves mitochondrial dysfunction in MM and inflammatory processes in PM/DM. Less prominent effects with the double-blind trial compared to the open-label trial were likely due to a lower amount of administered hydrogen and a shorter observation period, which implies a threshold effect or a dose-response effect of hydrogen. The complete article is available as a provisional PDF . The fully formatted PDF and HTML versions are in production.
关于硒元素(Selenium,Se)的生物学效应,研究得最多的恐怕是其抗癌作用,但学术界对此历来就存有争议 。 尽管如此,人们还是相信摄硒能够降低癌症风险。许多食物中都富含硒,如巴西坚果(brazil nuts,如下图)。然而,最近的一项随机控制临床试验( RCCT,randomized controlled clinical trials)研 究表明,硒对非黑素类的皮肤癌、前列腺癌都无防范作用。研究结果发表在 Cochrane Database Syst. Rev (2011年,第5期, DOI: 10.1002/14651858.CD005195 )上。主持这项研究的是来自德国柏林THR学院( Institute for Transdisciplinary Health Research )的 Gabriele Dennert 教授。 该研究组搜索了摄硒效应的临床研究文献,发现了49项预期的观察研究( observational studies )和6项随机控制临床试验。其中,观察研究表明,高硒摄入可以防范癌症,但RCCT研究却表明无此明显作用。另外还有迹象表明,如果摄硒时程过长,会造成有害效应。 Russo MW, Murray SC, Wurzelmann JI, Woosley JT, Sandler RS (1997). "Plasma selenium levels and the risk of colorectal adenomas". Nutrition and Cancer 28 (2): 125–9. doi : 10.1080/01635589709514563 . PMID 9290116 . Knekt P, Marniemi J, Teppo L, Helivaara M, Aromaa A (15 November 1998). "Is low selenium status a risk factor for lung cancer?" . American Journal of Epidemiology 148 (10): 975–82. PMID 9829869 . http://aje.oxfordjournals.org/cgi/pmidlookup?view=longpmid=9829869 . Young KJ, Lee PN (1999). "Intervention studies on cancer". European Journal of Cancer Prevention 8 (2): 91–103. doi : 10.1097/00008469-199904000-00003 . PMID 10335455 . Burguera JL, Burguera M, Gallignani M, Alarcón OM, Burguera JA (1990). "Blood serum selenium in the province of Mérida, Venezuela, related to sex, cancer incidence and soil selenium content" (Free full text). Journal of Trace Elements and Electrolytes in Health and Disease 4 (2): 73–7. PMID 2136228 . Clark LC, Combs GF, Turnbull BW, et al. (1996). "Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group". JAMA 276 (24): 1957–63. doi : 10.1001/jama.276.24.1957 . PMID 8971064 . Lippman SM, Klein EA, Goodman PJ, et al. (2009). "Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT)" (Free full text). JAMA 301 (1): 39–51. doi : 10.1001/jama.2008.864 . PMID 19066370 . "Chemoprevention Database" . http://www.inra.fr/reseau-nacre/sci-memb/corpet/indexan.html . Retrieved 2009-05-05 . Garland M, Morris JS, Stampfer MJ, et al. (1995). "Prospective study of toenail selenium levels and cancer among women". Journal of the National Cancer Institute 87 (7): 497–505. doi : 10.1093/jnci/87.7.497 . PMID 7707436 . Hercberg S, Galan P, Preziosi P, et al. (1998). "Background and rationale behind the SU.VI.MAX Study, a prevention trial using nutritional doses of a combination of antioxidant vitamins and minerals to reduce cardiovascular diseases and cancers. Supplementation en Vitamines et Minéraux Antioxydants Study" (Free full text). International Journal for Vitamin and Nutrition Research 68 (1): 3–20. PMID 9503043 . http://www.nlm.nih.gov/medlineplus/antioxidants.html . Hercberg S, Galan P, Preziosi P, et al. (2004). "The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals" (Free full text). Archives of Internal Medicine 164 (21): 2335–42. doi : 10.1001/archinte.164.21.2335 . PMID 15557412 . "Selenium and Chemotherapy - Nutrition Health" . http://nutrition-health.info/index.php/Selenium_and_Chemotherapeutic_Drugs . "Selenium Cancer" . http://nutrition-health.info/index.php/Selenium_Cancer_1 . Nilsonne G, Sun X, Nystrm C, et al. (2006). "Selenite induces apoptosis in sarcomatoid malignant mesothelioma cells through oxidative stress". Free Radical Biology Medicine 41 (6): 874–85. doi : 10.1016/j.freeradbiomed.2006.04.031 . PMID 16934670 . Tsavachidou D, McDonnell TJ, Wen S, et al. (2009). "Selenium and vitamin E: cell type- and intervention-specific tissue effects in prostate cancer" . Journal of the National Cancer Institute 101 (5): 306–20. doi : 10.1093/jnci/djn512 . PMC 2734116 . PMID 19244175 . http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrezartid=2734116 . Klein EA (2009). "Selenium and vitamin E: interesting biology and dashed hope". Journal of the National Cancer Institute 101 (5): 283–5. doi : 10.1093/jnci/djp009 . PMID 19244172 .
暂且先把以前的文章转来撑撑门面。 2008/11/17 20:48:30 晚上有牛同学的婚宴,五点半,食堂四楼。五点零七的时候刚要收拾东东走人,老板突然破门而入,头对头临床试验是怎么回事,帮我查查告诉我。 时间紧任务重啊! 网上搜刮了一下,好像没有定义,只能从提及的只言片语里寻找、推敲、归纳总结。在一篇介绍药物洗脱支架临床研究的文章里找到这样一段话,算是中文网页里说的比较明确的了,所谓直接比较研究即头对头(Head to Head)研究,又可分为优效性研究和等效/ 非劣效性研究;间接比较包括随机临床试验的亚组分析比较和注册临床研究的比较。( http://www.ccheart.com.cn/doctor/news/3702.html 《从循证医学角度看药物洗脱支架的临床试》)如此看来,头对头研究的目的主要是解决一般的临床试验中用亚组分析解决的问题,但解决的方法更直接按照特定的试验目的设计单独的临床试验,给药物的某些特殊特性的相关问题(如,治疗高血压的药物对靶器官的保护,干扰素在肝炎治疗中对不同基因型患者的疗效差别等)以直接明了的回答,也即针对药物(一般是疗效已经确定的药物)的特性设计完成单独的临床试验,进行临床研究,其目的已经不是研究药物是否有效,而是对药物疗效或安全性进行更加细致的研究,一般要求试验组与对照组的均衡性、可比性更强,使研究目的所针对的问题能够在尽可能单纯的条件下进行比较。《JIKEI HEART试验结果探析---联合抗高血压药物治疗》( http://www.bhli.org.cn/show.aspx?cid=10id=255 )中提到后期的临床试验大部分是头对头的试验,所谓头对头(Head to Head)研究即直接比较研究,又分为优效性研究和等效/非劣效性研究。试验不再简单讨论降压本身是否有益,而是要观察不同类别的降压药物在获得相同降压幅度的基础上,是否对靶器官有额外的保护作用。这样的临床试验常常需尽可能减小两组间血压变化的差别,使血压差异对结果的影响降低。由于降压治疗的临床获益主要来源于血压降低的幅度,因此,即使仅仅是平均2 mmHg~3 mmHg的血压差别的临床获益,已足以稀释了来自于药物本身的有益影响。这个例子就举得很好。 正当觉得已经比较明确的时候,又发现这样一个帖子,《何谓头对头试验》( http://www.med66.com/html/ziliao/yixue/7/cc078c6cc4d42df845e7c0e6782d9e29.htm )说就是单挑的意思, 呵呵, 比如两个药的效果的单挑。这下子还真有点蒙了,到底什么是头对头试验?看来只能找找英文网页了。于是乎首先想到去FDA的网站里搜搜看。 《The Healing Power of Placebos》( http://www.fda.gov/fdac/features/2000/100_heal.html )中提到:FDA doesn't require that a drug study include a placebo control group, DeLap says, only that its design be capable of establishing a drug's safety and effectiveness. Non-placebo types of drug studies include head-to-head studies, which compare the experimental drug to an existing treatment, and historically controlled studies, which compare the new drug's effects with information gathered in the past about the expected progression of a medical condition. 非常好,终于有比较确定的说法了,至少可以看出,头对头试验肯定是非安慰剂对照的试验,它是以临床上已经使用的治疗药物或治疗方法为对照的临床试验。 《Ottawa Panel evidence-based clinical practice guidelines for therapeutic exercises in the management of rheumatoid arthritis in adults.》( http://www.guideline.gov/summary/summary.aspx?ss=15doc_id=6264nbr=4019 )来自National Guideline Clearinghouse的一篇文章,其中提到Head-to-head studies (that is, the comparison of 2 active interventions, such as therapeutic exercises versus transcutaneous electrical nerve stimulation) were generally excluded in these recommendations. 由此可见所谓头对头研究就仅仅是指两种已经确认有效的治疗方法的比较。哎,没想到是这样,不是很甘心所以在一篇老外的帖子里提了下这个问题,等回信了再来补上最新消息吧。 头对头研究暂且告一段落。
最近在打击非法药品广告中, 有几位名人中了彩. 这几位公众人物是否做人厚道, 不是本来想关注的问题. 民众和舆论自有公道. 问题是这样的宣传, 商家为什么屡禁不止? 利益熏心是一方面原因, 另一方面是这种所谓名人效应和广告夸大宣传,还真管用. 老百姓信这一套. 就象某一治癌产品, 挂中科院技术名义, 整天在媒体做广告,夸大宣传,欺骗老百姓。 内部人员还很得意地说,如果不这样宣传,就没有人信,没有人买。这和抢钱骗钱有何区别? 中国是营养保健品泛滥成灾的国家。 有许多产品被夸大宣传。 吹得神乎其神。广告方面的规范和处罚很轻。那么在国外保健品,是否也有很多人信呢? 当然也是。 因为保健品的销售比药品松好多。国外的宣传攻势也很厉害。宣传多了,人们就信了。 商家也就等着数钱就是了。 关于营养保健品和复方维生素的功效, 在消费者心目中似乎是比较相信的, 不然不会卖得这么火爆. 这里有许多舆论宣传的功劳. 但医药界并非十分确信媒体造势所选宣传的诸多好处. 从循证医学的角度, 必须要有可靠的临床试验数据, 才能标上某种药效功能. 不然只能说未经证实和批准的疗效或个案报道. 最近纽约时报有一篇文章,引用了美国医学会刚发表的文章, 涉及银杏产品。 由于替代医学越来越越被人们所接受, 那么营养保健品或部分天然产物的临床疗效自然也受到业内的关注. 最新的数据是来自对银杏叶,这是在市场上流行的,广泛用于改善记忆和其他认知功能的天然产物或营养保健品。 研究者得到美国联邦政府资助,对3000多名年龄在72-96岁的老人进行随机分布,分别采取安慰剂或120毫克的银杏抽提物做成的制剂,一天服用两次。然后进行长期观察。 研究开始时,这些受试验的老人在服用安慰剂或银杏制剂时并没有患有痴呆症。所有受试验的人都经历中位数为6年的观察。 结果发现,那些服用银杏的人在记忆,语言,注意力和认知功能等测量指标上表现得并不比那些服用安慰剂的人更好。该成果发表在本周的美国医学会JAMA 杂志上,一个更早所完成的临床研究分析还发现,银杏没有减少人们患老年痴呆症的风险。 正如今年早些时候,纽约时报专栏曾经指出,在过去几年。有大型临床研究表明,某些维生素药片和其他营养补品不改善健康方面的关键参数和结果。 一位研究过这些数据和结果的医生告诉纽约时报记者说,我感到疑惑,为何市民普遍忽略了这种十分规范的临床试验熟结果, 公众对服用维生素和营养保健品的信任度,尽管没有得到现有科学数据的支持,但是仍然无法扭转局面,义无反顾地继续服用在专业人士看来是无功效的产品。实在让人费解。政府或许没有什么可做的,因为即使是安慰剂效应,只要没有安全隐患,政府无权禁止营养保健品的畅销,这全看商家的宣传本事。 Ginkgo biloba for Preventing Cognitive Decline in Older Adults A Randomized Trial JAMA. 2009;302(24):2663-2670. ABSTRACT Context The herbal product Ginkgo biloba is taken frequently with the intention of improving cognitive health in aging. However, evidence from adequately powered clinical trials is lacking regarding its effect on long-term cognitive functioning. Objective To determine whether G biloba slows the rates of global or domain-specific cognitive decline in older adults. Design, Setting, and Participants The Ginkgo Evaluation of Memory (GEM) study, a randomized, double-blind, placebo-controlled clinical trial of 3069 community-dwelling participants aged 72 to 96 years, conducted in 6 academic medical centers in the United States between 2000 and 2008, with a median follow-up of 6.1 years. Intervention Twice-daily dose of 120-mg extract of G biloba (n=1545) or identical-appearing placebo (n=1524). Main Outcome Measures Rates of change over time in the Modified Mini-Mental State Examination (3MSE), in the cognitive subscale of the Alzheimer Disease Assessment Scale (ADAS-Cog), and in neuropsychological domains of memory, attention, visual-spatial construction, language, and executive functions, based on sums of z scores of individual tests. Results Annual rates of decline in z scores did not differ between G biloba and placebo groups in any domains, including memory (0.043; 95% confidence interval , 0.034-0.051 vs 0.041; 95% CI, 0.032-0.050), attention (0.043; 95% CI, 0.037-0.050 vs 0.048; 95% CI, 0.041-0.054), visuospatial abilities (0.107; 95% CI, 0.097-0.117 vs 0.118; 95% CI, 0.108-0.128), language (0.045; 95% CI, 0.037-0.054 vs 0.041; 95% CI, 0.033-0.048), and executive functions (0.092; 95% CI, 0.086-0.099 vs 0.089; 95% CI, 0.082-0.096). For the 3MSE and ADAS-Cog, rates of change varied by baseline cognitive status (mild cognitive impairment), but there were no differences in rates of change between treatment groups (for 3MSE, P =.71; for ADAS-Cog, P =.97). There was no significant effect modification of treatment on rate of decline by age, sex, race, education, APOE*E4 allele, or baseline mild cognitive impairment ( P .05). Conclusion Compared with placebo, the use of G biloba, 120 mg twice daily, did not result in less cognitive decline in older adults with normal cognition or with mild cognitive impairment. Trial Registration clinicaltrials.gov Identifier: NCT00010803
 标签: 临床试验
标签: 临床试验