# 编者信息 熊荣川 明湖实验室 xiongrongchuan@126.com http://blog.sciencenet.cn/u/Bearjazz I was wondering if there is a tree statistic that compares the topology of two trees which gives and indication of the degree of similarity or dissimilarity between the trees? 我想知道是否有一个树的统计方法,比较两棵系统发育树的拓扑结构,给出和指示树之间的相似或不同程度? See these functions below, dist.topo is in library(Ape) # check for similarity # do the two trees produced by MrBayes and raxML have the same topology? all.equal(tree1, tree2, use.edge.length = FALSE) # result = TRUE or FALSE. # calculate the distance between the two tree, using default method. ( Penny and Hardy � s 1985) dist.topo(tree1, tree2) # result = numeric measure of distance 请看 R 包 Ape 中的以下函数, dist.topo 查看相似性 分别由 MrBayes 和 raxML 构建的两棵树,其拓扑结构一样吗? 使用默认参数,计算两棵树的距离 结果显示量化的距离 There is also SH.test (Shimodaira-Hasegawa test) in package phangorn And the Icong index by de Vienne which only compares topology (See this website: http://www.ese.u-psud.fr/utilisateurs/devienne/index.html) R 语言包 phangorn 中有个 SH.test (Shimodaira-Hasegawa test) 函数,也可检验两棵系统发育树的相似性。 de Vienne 提出的 Icong 指数只比较拓扑结构,到以下网址进一步了解详情。 To complete this discussion, the package distory computes Billera et al.'s geodesic distance between two trees. 为了终结本讨论, R 包 distory 可以计算由 Billera 等提出的两棵树之间测地距离。 参考文献: 【 1 】 https://grokbase.com/t/r/r-sig-phylo/1133cx4fck/comparing-the-topology-of-two-trees 【 2 】注解: geodesic distance ,这个单词的中文翻译是测地距离,其实测地距离的意思就是在三维空间中,两点之间的最短路径,归根究底就是最短路径,在三维中间从一个点到另外一个点的路径有无数种,但是最短路径只有一条,那么这个最短路径的长度就是测地距离 geodesic distance 。 --------------------- 作者: HNU_wang_chao 来源: CSDN 原文: https://blog.csdn.net/hnu_wang_chao/article/details/78612479
勇敢尝试,反复尝试,获取知识的唯一途径 面对一个问题,我们试着从不同的角度,不同的条件,不同的方法,去尝试解决问题。也就是必须反复思考,认真思考,深入思考,想想是否有解决问题的办法,很可能在反复的思考过程中获得好的想法。但这还不是真正的知识,因为,还未经过测试检验。只有通过检验,特别是实践的检验,才有可能成为人类的共同知识。检验也不是一次通过,失败了再来,回过头来再思考,试着再试一次,直到问题解决。 一个理论到底是“鲜花”还是“毒草”,应该摆出来任人评说。就算是错误的“毒草”,毛主席说,那也是很好的“肥料”。 理论物理的最新例子:在过去的 45 年中, M 弦理论是理论物理学舞台上占主导地位的理论。然而,在成千上万的世界上最好的物理学家们长达 45 年的努力后, M 弦理论还是濒临被抛弃。 在 2015 年十二月,在慕尼黑召开了专门讨论 M 弦理论的未来(为什么信任理论?)的会议。 Carlo 博士(量子重力组,中心主任,德吕米尼)会议上指出, M 弦理论完全在它自己的标准下未能完成使命。看 https://videoonline.edu.lmu.de/en/node/7477 M弦理论的验证标准由 M 弦理论本身确立的使命来检验: 一,其首要任务:计算标准模型的参数,(失败了) 二,推出得到三代粒子家族的存在,(失败了) 三,了解黑洞的最终命运,(失败了) 四,预测的宇宙学常数的符号,(失败了) 五,预测在 LHC 能发现新粒子,(失败了) 六,预测新的现象(黑洞与大型强子对撞机),(失败了) 七,预测牛顿引力的偏差在亚毫米的范围,(失败了) 八、预测低能量的超对称粒子,(失败了) 另一个非常杰出的理论物理学家西格尔写的一篇文章:“为什么 M 弦理论是不科学的理论”,请看: https://medium.com/starts-with-a-bang/why-string-theory-is-not-a-scientific-theory-9b3b2c2723ed#.xiw4f73ct 另一个非常杰出的物理学家 Peter Woit 的观点是: M 弦理论只是炒作概念,见: http://www.math.columbia.edu/~woit/wordpress/?p=8098 还有一位被称为现代的爱因斯坦, Lee Smolin 是美国著名理论物理学家。在他的畅销书, 《 物理学面临的问题:弦理论的兴起,科学的没落,和未来 》 讨论 M 弦理论中的面临一些具体问题,请看: https://en.wikipedia.org/wiki/The_Trouble_with_Physics 显然, M 弦理论问题如此多多,但这是人类尝试解释自然世界的一次勇敢的尝试。虽然经过 45 年多的奋斗和努力, M 弦理论目前濒临死亡,但所有的物理学家都赞赏和感激 M 弦理论: 一,尽管 M 弦理论还没有作为一个成功物理理论的事实,它产生了许多伟大的副产品。 M 弦理论的关键发明人,美国理论物理学家、数学物理学教授,在普林斯顿、新泽西高等研究院工作的爱德华已获得“菲尔茨数学奖”等奖项。看: https://en.wikipedia.org/wiki/Edward_Witten 二, M 理论有一个非常漂亮的数学结构。但是,它的有效性只能通过自然的判断。西方人的高明之处在于西方人不会假装他们知道这一切,并且可以作为掌管自然秘密的判官。 45 年了,他们没有禁止 M 弦理论的传播。虽然 M 弦理论本身没有创造出一个真实可信的物理理论。今天,我们仍然感谢他们开放的心态,我们从中学到了很多东西。 通过 M 弦理论的尝试失败,但让我们知道那个是死胡同,因此我们会更聪明。这就是一个伟大的知识探索的尝试。 可以这样说, M 弦理论这棵“毒草”,作为很好的肥料,滋养了一、二代人,而且,还培育了世界上最美丽的“花朵”。这个花朵还没有被人看见,但我们知道它存在。 所以,尝试,尝试,不断尝试,是获得知识的唯一途径。任何禁止尝试的举动,那是无知的万全之策,阻挡着科学探索的成功之路。 正是: 花花草草舞缤纷,万紫千红才是春,创新总要多尝试,求得仙草救病人。
广义 Kulback-Leibler 语义信息公式和最大似然法的一致性 鲁晨光 (这是一篇长文的摘要,删除了部分哲学讨论,保留了和统计及信息论相关的内容,目的是让研究最大似然法的学者看看。我相信文中广义信息公式可以比Kullback-Leibler公式更方便表达和解决最大似然问题,并能解决最大似然学派和贝叶斯学派的矛盾。文中公式(15)是一个重要结论,应该能给最大似然估计供极大方便。笔者研究估计问题时间不长, 不妥之处欢迎指正。) 1. 引言 Shannon ( 1948 ) 发表文章:《通信的数学理论》 ,随后 Weaver 提出语义信息 研究方向, Bar-Hillel, Y. 和 Carnap ( 卡尔纳普 ) 提出用逻辑概率代替统计概率度量语义学信息 . 公式是 inf( i )=-log m p ( i ) 。其中 i 是命题, m p 是逻辑概率。不过最早提出概率和信息反相关的却是 Popper ( 波普尔 ) 。 Popper 早在 1935 年的著作《科学发现的逻辑》 ( , 96,269 ) 中就提出用可检验性,或可证伪性,或信息作为科学理论划界和评价的准则,并且明确提出,概率越小,信息量越大。后面谈到, Popper 提出的检验的严厉性公式 ( ,526) 稍加改善,就可以用作语义信息计算。 在 Barhillel , Carnap 和 Popper 之后, 西方语义信息测度的研究总结见 , 关于信息哲学的研究总结见 。西方最有代表性的研究者是 Floridi 。中国最著名的语义信息倡导者和研究者是钟义信 . 另外也有其他学者研究广义信息 或多或少联系到语义信息。但是根据上述研究,我们仍然不能计算一个简单预测比如 “ 明天有大雨 ” 或 “ 小偷大约 20 岁 ” 的信息;或 GPS 箭头、手表指针、温度表和秤的读数提供的信息。 另一方面,自从 Akaike 把 Fisher 的最大似然度方法和 Kullback-Leibler ( 后面简记为 KL ) 公式联系起来讨论估计的优化,越来越多的归纳问题研究者意识到,最大似然度方法信息方法相结合可以同时解释证伪和归纳 。他们的研究已经把我们带到迷宫入口附近。但是如何根据事实发生的样本序列确证一个预测,比如 “ 明天有大雨 ” , “ 所有天鹅是白的 ” ,并算出它们的确证度? 依然众说纷纭,没有一致结论 。 笔者以为,流行的语义信息和归纳问题研究的困难都是由于:统计概率,逻辑概率,命题真值,真值函数等没有很好区分,比如同时用 P 表示统计概率和逻辑概率,同时用 E 表示个体和变量,因而使得分析的框架不清晰。 笔者曾提出和 Shannon 及 Popper 理论兼容的广义信息论 ,它能很好解释证伪。笔者最近研究发现,可以通过降低对假设的信任度,减少预测失误带来的信息损失,提高平均信息。这样,平均语义信息公式就可以同时用于计算 Popper 的信息和优化现代归纳主义研究的确证度。和流行的做法不同,这个公式同时使用了逻辑概率 ( 用 T 表示 ) 和统计概率 ( 用 P 表示,反映证据和背景知识 ) 。重要的是,公式还使用了模糊真值函数 ( 即条件逻辑概率 ) 以及信任度 c ( 它在 -1 和 1 之间变化 ) 。本文继承或关系到 Popper , Shannon , Barhil and Carnap, Zadeh , Kulback and Leibler , Fisher , Akaike 等人的研究结果。 下面首先讨论谓词的真值函数和逻辑概率,以及它们和统计概率之间的关系。然后通过推广经典信息公式得到平均语义信息公式和广义 Kullback-Leibler 公式,说明它们如何用于预测的信息评价,如何符合 Popper 用于检验或证伪的信息准则。文中最后讨论,如何优化假设,包括优化信任度 c ,从而提高平均语义信息,使之达到其上限: KL 信息。 2. 真值函数和逻辑概率 2.1 命题的真值和谓词的真值函数 日常语言中,语句真假往往是模糊的。比如猜测 “ 小偷大约 20 岁 ” ,这话的真假是模糊的,该在 0 和 1 之间变化。如果小偷真的 20 岁,预测真值就是 1 ,如果有偏差, 比如是 25 岁, 真值就变小, 比如说是 0.5 ;如果是 30 岁,真值就更小。所以日常语言的真值函数取值于实数区间 而不是二值集合 {0,1}. 后面讲到的真值函数都是模糊真值函数。 我们用大写字母 E 表示一个变量, 代表一个个体 ( individual ) 或证据,它是个体 e 1 , e 2 ,… , e m 中的一个,这些个体构成集合 A , 于是有 E ∈ A ={ e 1 , e 2 , … , e m } 。 E = e i 表示 e i 发生。类似地, 预测或假设是 H ∈ B ={ h 1 , h 2 , …, h n }. 一个预测 h j 发生后, E = e i ,预测就变为命题 h j ( e i ). 用经典信息论的语言来说, P ( E ) 是信源或先验概率分布, P ( H ) 是信宿。条件概率矩阵 P ( H | E ) 是信道。对于语义通信来说,在 Shannon 信道之外还存在语义信道 T ( H | E ) 。 一个典型的语义通信例子是天气预报, E 表示降水量,比如 15mm. H 表示降水量预报。 比如 h 1 = “ . 无雨 ” ( 比如 “ 明天无雨 ” , 其他类推 ) , h 2 = “ . 有雨 ” , h 3 = “ . 小雨 ” , h 4 = “ . 中雨 ” , h 5 = “ . 小到中雨 ” … H = h j 表示 h j 被选择。类似的例子是关于年龄 ( E ) 的一组陈述 ( H ) : “ . 是小孩 “ , “ . 是年轻人 ” , “ . 是中年人 ” , “ . 是老年人 ” 。 另一个典型的语义通信方式是数值预测或估计 ( 后面简称估计,数学上通常记为 e^ j , e^ j = h j = h j ( E )= “ E ≈ e j ”= “ E 大约是 e j ” 。不光是语言表达的估计, GPS 的箭头, 手表的指针,甚至一种色觉, 都可以看做是一个估计 . 估计的例子参看表 1. 表 1 估计 h j = e^ j = “ E ≈ e j ” 举例 例子 预测或假设 h j = “ E ≈ e j ” 事实或证据 E e i h j ( e i ) 的真值 T ( A j | e i ) 大约是 日常语言 “ 小偷大约 20 岁 ” 小偷实际年龄 18 岁 0.9 经济预测 “ 今年股市可能涨 20% ” 实际涨幅 0 0.1 秤 秤的读数 “ 1KG ” 实际重量 0.9KG 0.3 GPS 地图上箭头 ↖ 实际位置 偏右 5 米位置 0.9 色觉 一种色觉比如黄色觉 实际色光,带有某种主波长 主波长是 660nm 的色光 0.7 用 Zadeh 开创的模糊数学的语言说,相对 h j = h j ( E ), A 中有一个使 h j 为真的模糊子集 A j , 一个元素 E 在 A j 上的隶属度函数 m Aj ( E ) 就是就是 h j 的真值函数, 记为 T ( h j ( E ))= T ( h j | E )= T ( A j | E )= m Aj ( E ) (1 ) 当 E = e i 时,真值函数就变为真值 T ( A j | e i ). 天气预报等自然语言的真值函数来自习惯用法,后面将证明它们来自过去的条件概率函数 P ( h j | E ) 。如果不知道过去的 P ( h j | E ) ,也可以采用随机集合的统计方法得到 。而估计 h j = “ E ≈ e j ” 的真值函数来自人工定义和实际误差概率分布 —— 也取决于过去的条件概率 P ( h j | E ) ,可以近似地用指数函数 ( 没有系数的正态分布 ) T ( A j | E )=exp ( 2 ) 表示,其最大值是 1 。其中 d 表示标准差, 反映估计的模糊程度, d 越大,估计就越模糊 , 函数波形覆盖面积越大。这里我们假设这些估计都是无偏估计,有些非无偏估计可以通过对 E 的转换得到, 比如用 E 0.5 代替 E ,使估计成为无偏估计。 假设相对每个 h j 或 A j , 存在一个 e j ( 相当于柏拉图的理念和我让通常说的典型 ) 使得 T ( A j | e j )=1, 那么, h j ( e i ) 的真值 T ( A j | e i ) 就可以理解为 e i 和 e j 的相似度或混淆概率。 2.2 逻辑概率 T ( A j ) 及其和真值函数 T ( A j | E ) 及信源 P ( E ) 的关系 后面内容见附件 语义信息最大似然度理论-short博文.pdf
编者(熊荣川)按:正选择检测是指通过检测编码基因序列变化(或进化)中的非同义突变和同义突变的差异来量化进化压力对遗传变异的贡献;可用于推断基因的功能性或者功能位点,是一种常用的基因分析及筛选手段,下面是一段文摘,对如何进行有效的正选择检验进行了较为详细的阐述,具有较高的参考价值。 Positiveselection was tested using the REL(HyPhy) and CODEML (PAML) methodsand these analyses revealed that codons exhibiting high dN/dS ratios, andtherefore likely to have been subjected to positive selection, were enriched inthe N-terminal cytoplasmic and TM domains in primate tetherins (Fig. 7).Tetherin evolution in primates was also evaluated under several standard modelsof sequence evolution as implemented in the CODEML program. These comprisethree nested pairs of models (M0 and M3; M1a and M2a; M7 and M8) in which thesecond model of each pair is derived from the first by allowing sites to evolveunder positive selection. Nested models were compared using the likelihoodratio test, and in each case allowing individual sites to evolve under positiveselection (M3, M2a, M8) gave a significantly better fit to the primate sequencedata than the corresponding model without positive selection (M0, M1a and M7,respectively) (Table 1). The M3, M2a and M8 models identified a largelyoverlapping set of sites in the tetherin coding sequence with dN/dS.1,consistent with an evolutionary history characterized by frequent episodes ofpositive selection. Notably, some codons that exhibited a high probability ofhaving evolved under positive selection coincided with residues that determinedthe effectiveness of Vpu antagonism (Fig. 7). However, there were numerousadditional codons, particularly in the tetherin cytoplasmic domain, that alsoexhibited high dN/dS ratios, suggesting that antagonists other than Vpu havealso imposed selective pressure on primate tetherin sequences. 参考文献 McNatt, M. W., T.Zang, T. Hatziioannou, M. Bartlett, I. B. Fofana, W. E. Johnson, S. J. Neil andP. D. Bieniasz (2009). Species-specific activity of HIV-1 Vpu and positiveselection of tetherin transmembrane domain variants. PLoS pathogens 5 (2): e1000300.
转自 http://blog.renren.com/share/227263222/15891121172?from=0101010202ref=hotnewsfeedsfet=102fin=22fid=22617737969ff_id=227263222platform=0expose_time=1370234440 数理统计学了这么多年,唯一的感觉就是没学透。很多东西搞不清楚,检验(testing)就是其中一项。提到检验,脑子里浮现出来的就是T检验,NP引理,显著性水平,置信区间什么的,不成体系。记得王老师有讲过显著水平的意义,可是当时并未完全理解,也就一直这么糊里糊涂的过来了。2005年的The American Statistician中有一篇文章,题为Testing Fisher, Neyman , Pearson, and Bayes. 这篇文章描述了不同检验的逻辑脉络,对于有一定数理统计学基础,但是有没有研究的那么深刻,比如我这样的童鞋来说,可以说是醍醐灌顶的。文章不长,只有六页,感兴趣的童鞋可以自己上网搜搜看看。下面写点读文章的体会。 统计学界最有名的争论之一,是Fisher与Neyman-Pearson之间关于如何进行检验之间的争论。另外一个最有名的争论,则是Fisher和巨大多数贝叶斯学派之间的争论。这篇文章,则是通过一个简单的例子,来展示不同的检验方法。结论是Fisher的检验与NP检验,Bayes检验不可比,原因是它检查的是一个不同的问题。而Bayes检验,则相对NP检验更加好。 Fisher的检验的一些基本的想法是: (1)对于数据,有个概率模型。 (2)对于多维的数据,有一个分布已知的统计量。 (3)已知的分布能够给那些奇怪的值进行排序。 (4)p值,用来评估奇怪的程度。 (5)alpha显著性水平是p值用来参考的。 NP检验的一些基本的想法是: (1)对于数据,有两个可供选择的模型。 (2)alpha值是原假设为真时拒绝原假设的概率。 (3)拒绝域的选择适应了alpha值的定义。 (4)有很多基于功效函数的检验模式。 (5)在比较复杂的为体重,无偏性和不变性会用来约束功效函数。 而Bayes则引入了关于分布参数的先验信息,经过贝叶斯公式计算得到参数的后验概率,然后通过后验概率分布情况,类似的检验。检验的想法与之前介绍的无异。只是引入了先验信息的想法。 在这篇文章的最后,作者提到,他们在教回归的时候,抛弃了NP想法,这个老师可能是Bayes学派的,他说”In theory courses I teach some NP testing because of its historical role and the face that other statisticians expect student to know it. If I could get away with it, I would teach introductory statistics from a Bayesian point of view” 。 American statistician这个杂志之前并没有听说过,其中有很多讨论关于统计教学问题的文章。有些老师的文章对于一些比较基础的统计理论阐释的很清楚,对于我等伪学术者,看这样的杂志感觉很好。 下面是我今天无聊,翻译的文章的主要部分。夏日炎炎,心情非常烦躁,写点东西,心里还踏实点。 -------------------------------------------------------------------------------------------------------------------------------------- 统计学界最有名的争论之一,是Fisher与Neyman-Pearson之间关于如何进行检验之间的争论。另外一个最有名的争论,则是Fisher和巨大多数贝叶斯学派之间的争论。这篇文章,则是通过一个简单的例子,来展示不同的检验方法。结论是Fisher的检验与NP检验,Bayes检验不可比,原因是它检查的是一个不同的问题。而Bayes检验,则相对NP检验更加好。 下面通过一个简单的例子来看看这些不同的检验自身的逻辑基础以及他们之间的异同。 1. Fisherian Tests “Fisher的检验”这个称呼完全是这篇文章的作者为了叙述方便给起的。Fisher自己并没有给他提出的检验方法起这样一个名字。事实上,Fisher的检验,其本质是对显著性的检验(tests of significance),相比之下,NP检验则是一种对假设的检验(tests of hypotheses)。 从名字的不同,我们可以看出一些端倪。Fisher的检验,完全不会涉及到备择假设。(alternative hypothesis),这种检验可以作为一种模型验证过程。其逻辑是这样的:我们有了一个模型的分布情况,然后我们检查我们的数据对于这个模型看起来是不是很奇怪。 我们举个简单的例子来说明这个事。随机变量可以取四个值r=1,2,3,4,随机变量的分布则取决于参数θ,θ可以取值0,1,2.分布情况如下表所示。 若要检验的假设为H0:θ=0,则Fisher的检验是完全基于如下的分布情况的: 有了分布情况,我们就可以看出哪些值很奇怪,而哪些值不是。比如在这个例子中,如果我们的显著性水平alpha=0.01,显然,奇怪的观测值就为r=2,3,然后是r=4。对于显著性水平alpha=0.01的检验,我们在观测到2或者3的时候拒绝这个模型,当alpha=0.02时,我们观察到2,3,4的时候拒绝模型。 为了替代显著性水平alpha,Fisher用p值来评估检验。所谓的p值,是指看到奇怪的观测的概率,或者比你看到的更奇怪的概率。在我们的例子中,最奇怪的观测是2和3,它们俩是一样奇怪的,因此p值是0.01(0.05+0.05)。如果你观察到了4,那么2和3都是比4更奇怪的,因此p值是0.02(0.1+0.05+0.05) 在Fisher的检验中,p值是比alpha值更为基本的概念。从技术上讲,alpha值仅仅是一个进行选择的准则,换句话讲,alpha只是一个选择的临界点,来判断那些观测是奇怪的,那些是不奇怪的。如果p值小于alpha,则拒绝我们的原假设的模型。 Fisher检验的逻辑基础是通过反例来进行证明。我们假设一个模型,然后我们通过观测到的样本来检查我们的假设,如果观测很奇怪,对于我们的假设模型,这样的观测是非常不可能出现的,那么我们就拒绝我们的假设。P值则给出了一个度量,来度量数据与模型并不矛盾的程度。(p值越大,与假设的模型越不矛盾)。 对于一个通过反例来证明的过程,结果往往可能会被误解。如果数据与模型矛盾,我们就有证据来拒绝模型,但是如果数据与模型并不矛盾,我们或许可能试图证明模型是正确的。但是从逻辑上讲,我们只是还没有证明我们的模型是错误的,从而拒绝他,但是并不能证明正确性。不拒绝,是一种态度,并不是接受,而是仅仅因为没有证据能够证明模型不好。永远不要去接受一个原假设模型。 上面以及提到,Fisher检验,并不需要备择假设。但是NP检验和Bayes检验,则都需要备择假设。我们下面通过不同的备择假设,来分别介绍NP和Bayes检验的逻辑。 2. SIMPLE .V.S .SIMPLE 在学习数理统计假设检验的过程中,首先遇到的假设检验就是简单对简单的。比如,H0:θ=0,H1:θ=2。这样,我们的检验问题,其实就变成了一个选择问题,我们有两个选项,我们要从中选择一个。Fisher反对NP检验的原因,也最初是基于此的。 对于这个检验问题,我们需要的信息如下 在进行正式的假设检验的过程之前,我们先来看看这个问题的分布情况。如果我们看到r=4,我们就比较倾向于认为θ=2,而我们如果看到r=1,则我们倾向于认为θ=0,如果我们看到r=2或者3,我们依然可以认为数据来自于θ=0这个模型的可能性(注意这里的措辞)是五倍于θ=1这个模型。 虽然Fisher的检验并不涉及到备择假设,但是我们可以对两个假设分别进行Fisher的检验,然后比较检验的结果。对于假设θ=0,r=2,3,4时候都有比较小的p值,而对于假设θ=2,r=2,3的时候p值比较小。当r=4的时候,我们不能拒绝θ=2;当r=1的时候,我们不能拒绝θ=0;当r=2,3的时候,两个假设我们都可以拒绝。Fisher的检验,并不强迫我们选择一个备择假设。 2.1. Neyman-Pearson Tests NP检验,对于两个假设并不是同等对待的。检验问题H0:θ=0.VS.H1:θ=2往往和检验问题H0:θ=0.VS.H1:θ=2是不同的。我们检验前一个问题。 NP定理是为了寻找一个最好的显著性水平alpha,alpha是当原假设为真是拒绝原假设的概率。所谓的拒绝域,是指能够拒绝原假设的观测集,因此在原假设下,拒绝域的概率一定是alpha。所谓最好的检验,就是具有最高功效的检验,也就是当备择假设为真的时候,拒绝原假设的概率最高。 为了拒绝原假设而确定alpha的过程,强调有重复的抽样,因此大数定律告诉我们,大约有alpha次我们的选择会是错误的。为了理解显著性水平alpha的抽象意义,我们必须要考虑到随机化检验(randomized tests)。随机化检验的拒绝域是随机化的。举个例子来说,我们如何进行一个显著性水平alpha=0.0125的检验?三种不同的检验如下: a) 只要r=4就拒绝,然后抛硬币,如果正面向上,那么当r=2的时候拒绝。 b) 只要r=4就拒绝,然后抛硬币,如果正面向上,那么当r=3的时候拒绝。 c) 只要r=2或者3,就拒绝,然后抛硬币两次,如果两次都是正面朝上,则当r=4的时候拒绝。 但是这样的过程是实践起来是很难说服人的。 NP引理,告诉我们最优的NP检验是基于似然比的,f(r|2)/f(r|0)。似然比值最大时,最优的NP检验给出拒绝的结果,因此alpha=0.01时,NP检验当r=4的时候是拒绝的。这个和Fisher的检验是完全不同的。(Fisher的检验当alpha=0.01时,是在r=2,3的时候拒绝)。对于显著性水品为0.01的NP检验而言,功效是0.9,而对于同样显著性水平的Fisher检验,功效则为0.01+0.01=0.02。很显然,对于有备选的情况下,Fisher的检验并不是特别适用,因为Fisher的检验本身就不是为了这种问题而设计的。对于Fisher的检验和NP检验而言,alpha的概念是不同的。这两种检验之间并没有可比之处。了解了两种检验的逻辑基础,我们就可以有选择的选择,从而适用某些特定的问题。 下面的例子,让我们看到p值,在NP检验中没有起到什么作用。 比如检验问题是H0:θ=1.VS.H1:θ=2,则最优的NP检验当r=4的时候拒绝,然而,在原假设下,r=4的概率是0.5,是最可能被观测的值。这说明NP检验的逻辑基础,并不是通过反例来证明,而且整个检验的过程中,确实没p值什么事儿。 另外,值得注意的是,原假设与备择假设的选择也是非常重要的,你需要选择哪个假设作为原假设,这也是一个选择的过程。即便如此,NP检验表现的也并不怎么好。再举一个例子,显著性水品alpha=0.02的NP检验H0:θ=0.VS.H1:θ=2的拒绝域包括了r=2,3,但是2,3在原假设下也比备择假设下看上去出现的可能性要大四倍。在两种假设下,2,3都是比较奇怪的观测,但是我们在这两个假设中进行选择,当r=2,3的时候拒绝了θ=0,从而接受了θ=2,这看起来并不合理。而下面要介绍的贝叶斯检验,则比较好的解决了这个问题。 2.2. Bayesian Tests Bayes就是要求我们对于参数θ有先验的概率分布。然后利用Bayes定理,将先验和当前的观测结合在一起,得到θ的后验概率分布。然后所有的关于θ的决策都建立在后验概率分布的基础上。而数据中的信息是从似然函数中来的。 在我们这个简单对简单的例子中,我们假设θ=0,2的先验概率分别为p(0),p(2),然后利用Bayes定理,我们就可以从数据中得出后验概率,记为p(0|r),p(2|r)。 具有较大后验概率的值将被接受。如果两个假设的后验概率都差不多,那么我们只能承认,我们也不知道那个假设是正确的了。Bayes检验的特点在于,它公平的对待了两个假设,不区分所谓的原假设与备择假设。 但是Bayes最引人争议之处,在于先验概率的确定。检验过程往往缺乏比较明确的先验信息。但是,如果我们的数据足够多,那么先验信息的影响力可能并不那么大。而如果我们的数据不那么充足,则先验信息影响很大,不同的先验信息得到的检验结果并不相同,但是我们为什么要期望检验的结果是相同的呢?在这个例子中,只要有r=1或者4这样一次观测就足以使我们作出决策,而如果观测是2或者3,则说明我们需要更多数据。 3. SIMPLE VERSUS COMPOSITE 我们现在考虑进行这样的检验H0:θ=0,H1:θ0。我们的例子非常简单,直接观察表一,我们就能得到一些比较明确的结论。比如r=1时候,我们倾向于认为θ=0,r=4的时候,倾向于θ=2,r=2,3的时候,则倾向于θ=1。 对于这样的检验问题,Fisher的检验并没有什么特别值得说明之处。 而对于NP检验而言,我们期望得到的是均匀的最有功效的检验(uniformly most powerful test)。我们取θʹ,是一个大于0的值,然后我们把检验问题重新转化为简单对简单的 H0:θ=0,H1:θ=θʹ。如果对于对于给定的显著性水平alpha,最有功效的检验得到的拒绝域是相同的,而不考虑θʹ的值,这样的检验就被称为均匀的最有功效检验。比如说,对于alpha=0.01,r=4的时候拒绝就是均匀的最有功效检验。 没有偏见的贝叶斯检验,需要认为原假设和备选假设的出现概率是相同的,也就是说,在这个简单对复杂的问题中,先验信息为p(θ=0)=0.5,p(θ0)=0.5,然后就是利用贝叶斯公式计算后验概率如下: 这个后验概率分布情况,相对来说对于检验就比较明确了。
rank sum test 秩和检验 秩和检验方法最早是由维尔克松(Wilcoxon)提出,叫维尔克松两样本检验法。后来曼—惠特尼将其应用到两样本容量不等(n1不等于n2)的情况,因而又称为曼—惠特尼U检验。这种方法主要用于比较两个独立样本的差异。 1、假设中的等价问题 设有两个连续型总体, 它们的概率密度函数分别为: f 1 ( x ), f 2 ( x )(均为未知) 已知 f 1 ( x ) = f 2 ( x − a ),a为末知常数,要检验的各假设为: H 0 : a = 0, H 1 : a 0. H 0 : a = 0, H 1 : a 0. H0:a=0,H1, a0. 设两个总体的均值存在,分别记为μ 1 ,μ 2 ,由于 f 1 , f 2 最多只差一平移,则有μ 2 = μ 1 − a 。此时, 上述各假设分别等价于: H 0 :μ 1 = μ 2 , H 1 :μ 1 μ 2 H 0 :μ 1 = μ 2 , H 1 :μ 1 μ 2 H 0 :μ 1 = μ 2 , H 1 :μ 1 μ 2 2、秩的定义 设X为一总体,将容量为n的样本观察值按自小到大的次序编号排列成 x (1) x (2) Λ x ( n ) ,称 x ( i ) 的足标i为 x ( i ) 的秩, i = 1,2,Λ, n 。 例如: 某施行团人员的行李重量数据如表: 重量(kg) 34 39 41 28 33 写出重量33的秩。 因为2833343941,故33的秩为2。 特殊情况: 如果在排列大小时出现了相同大小的观察值, 则其秩的定义为足标的平均值。 例如: 抽得的样本观察值按次序排成0,1,1,1,2,3,3, 则3个1的秩均为(2+3+4)/3=3. 两个3的秩均为(6+7)/2=6.5. 3、秩和的定义 现设1,2两总体分别抽取容量为 n 1 , n 2 的样本,且设两样本独立。这里总假定 n1n2。 我们将这 n 1 + n 2 个观察值放在一起,按自小到大的次序排列,求出每个观察值的秩,然后将属于第1个总体的样本观察值的秩相加,其和记为 R 1 ,称为第1样本的秩和,其余观察值的秩的总和记作 R 2 ,称为第2样本的秩和。 显然, R 1 和 R 2 是离散型随机变量,且有 R 1 + R 2 =( (n1+n2)(n1+n2+1) )/2. 4、秩和检验法的定义 秩和检验是一种非参数检验法, 它是一种用样本秩来代替样本值的检验法。 用秩和检验可以检验两个总体的分布函数是否相等的问题 秩和检验的适用范围 如果两个样本来自两个独立的但非正态获形态不清的两总体,要检验两样本之间的差异是否显著,不应运用参数检验中的 T检验 ,而需采用秩和检验。 秩和检验的方法 1、两个样本的容量均小于10的检验方法 检验的具体步骤: 第一步:将两个样本数据混合并由小到大进行等级排列(最小的数据秩次编为1,最大的数据秩次编为 n 1 + n 2 )。 第二步:把容量较小的样本中各数据的等级相加,即秩和,用T表示。 第三步:把T值与秩和检验表中某α显著性水平下的临界值相比较,如果 T 1 T T 2 ,则两样本差异不显著;如果TT1或T=T2, 则表明两样本差异显著。 例: 某年级随机抽取6名男生和8名女生的英语考试成绩如表1所示。问该年级男女生的英语成绩是否存在显著差异? 男、女生英语考试成绩表 解: 检验步骤: (1)建立假设: H 0 :男女生的英语成绩不存在显著差异 H 1 :男女生的英语成绩存在显著差异 (2)编排秩次,求秩和: T= 13 + 7 + 14 + 12 + 5.5 + 11= 62.5 (3)统计推断:根据 n 1 = 6, n 2 = 8,α = 0.05, 查秩和检验表,T的上、下限分别为 T 1 = 29, T 2 = 61,有 T T 2 ,结论是:男女生的英语成绩存在显著差异。 3、两个样本的容量均大于10的检验方法 当两个样本容量都大于10时,秩和T的分布接近于正态分布,因此可以用Z检验,其基本公式为: 式中:T为较小的样本的秩和。 例: 某校演讲比赛后随即抽出两组学生的比赛成绩如表2,问两组成绩是否有显著差异? 解: 检验步骤: (1)建立假设: H 0 :两组成绩不存在显著差异 H 1 :两组成绩存在显著差异 (2)编排秩次,求秩和: n 1 = 12, n 2 = 14, T = 144.5,代入公式,有: (3)统计推断:因为|Z|1.96,则应保留虚无假设,拒绝备择假设。结论是:两组的演讲比赛成绩不存在显著差异。
原文地址:http://blog.sina.com.cn/s/blog_5ecfd9d90100cigp.html 在统计学中,柯尔莫可洛夫-斯米洛夫检验基于累计分布函数,用以检验两个经验分布是否不同或一个经验分布与另一个理想分布是否不同。 In statistics , the Kolmogorov – Smirnov test (K–S test) is a form of minimum distance estimation used as a nonparametric test of equality of one-dimensional probability distributions used to compare a sample with a reference probability distribution (one-sample K–S test), or to compare two samples (two-sample K–S test). The Kolmogorov–Smirnov statistic quantifies a distance between the empirical distribution function of the sample and the cumulative distribution function of the reference distribution, or between the empirical distribution functions of two samples. The null distribution of this statistic is calculated under the null hypothesis that the samples are drawn from the same distribution (in the two-sample case) or that the sample is drawn from the reference distribution (in the one-sample case). In each case, the distributions considered under the null hypothesis are continuous distributions but are otherwise unrestricted. The two-sample KS test is one of the most useful and general nonparametric methods for comparing two samples, as it is sensitive to differences in both location and shape of the empirical cumulative distribution functions of the two samples. The Kolmogorov–Smirnov test can be modified to serve as a goodness of fit test. In the special case of testing for normality of the distribution, samples are standardized and compared with a standard normal distribution. This is equivalent to setting the mean and variance of the reference distribution equal to the sample estimates, and it is known that using the sample to modify the null hypothesis reduces the power of a test. Correcting for this bias leads to the Lilliefors test . However, even Lilliefors' modification is less powerful than the Shapiro–Wilk test or Anderson–Darling test for testing normality. Kolmogorov–Smirnov statistic The empirical distribution function F n for n iid observations X i is defined as where is the indicator function , equal to 1 if X i ≤ x and equal to 0 otherwise. The Kolmogorov–Smirnov statistic for a given cumulative distribution function F ( x ) is img class="tex" alt="D_n=\sup_x |F_n(x)-F(x)|," src="http://upload.wikimedia.org/math/3/b/8/3b8599f003f2a131d8084621b1c39640.png" real_src="http://upload.wikimedia.org/math/3/b/8/3b8599f003f2a131d8084621b1c39640.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / where sup S is the supremum of set S . By the Glivenko–Cantelli theorem , if the sample comes from distribution F ( x ), then D n converges to 0 almost surely . Kolmogorov strengthened this result, by effectively providing the rate of this convergence (see below). The Donsker theorem provides yet stronger result. Kolmogorov distribution The Kolmogorov distribution is the distribution of the random variable img class="tex" alt="K=\sup_{t\in }|B(t)|," src="http://upload.wikimedia.org/math/1/b/7/1b7fd8f556e7382d973cb6bf95a245ea.png" real_src="http://upload.wikimedia.org/math/1/b/7/1b7fd8f556e7382d973cb6bf95a245ea.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / where B ( t ) is the Brownian bridge . The cumulative distribution function of K is given by img class="tex" alt="\operatorname{Pr}(K\leq x)=1-2\sum_{i=1}^\infty (-1)^{i-1} e^{-2i^2 x^2}=\frac{\sqrt{2\pi}}{x}\sum_{i=1}^\infty e^{-(2i-1)^2\pi^2/(8x^2)}." src="http://upload.wikimedia.org/math/2/8/9/2899bf257fc0aa1f48b3ffcff8f783ae.png" real_src="http://upload.wikimedia.org/math/2/8/9/2899bf257fc0aa1f48b3ffcff8f783ae.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / Kolmogorov–Smirnov test Under null hypothesis that the sample comes from the hypothesized distribution F ( x ), img class="tex" alt="\sqrt{n}D_n\xrightarrow{n\to\infty}\sup_t |B(F(t))|" src="http://upload.wikimedia.org/math/8/4/2/842d0b1d85ca11aa30ccc90a09936fa4.png" real_src="http://upload.wikimedia.org/math/8/4/2/842d0b1d85ca11aa30ccc90a09936fa4.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / in distribution , where B ( t ) is the Brownian bridge . If F is continuous then under the null hypothesis img class="tex" alt="\sqrt{n}D_n" src="http://upload.wikimedia.org/math/1/e/c/1ec425f3720cd63ffabd65504c798972.png" real_src="http://upload.wikimedia.org/math/1/e/c/1ec425f3720cd63ffabd65504c798972.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / converges to the Kolmogorov distribution, which does not depend on F . This result may also be known as the Kolmogorov theorem ; see Kolmogorov's theorem for disambiguation. The goodness-of-fit test or the Kolmogorov–Smirnov test is constructed by using the critical values of the Kolmogorov distribution. The null hypothesis is rejected at level α if img class="tex" alt="\sqrt{n}D_nK_\alpha,\," src="http://upload.wikimedia.org/math/8/9/1/891bbf7487bdbedcc202cb47bee880ac.png" real_src="http://upload.wikimedia.org/math/8/9/1/891bbf7487bdbedcc202cb47bee880ac.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / where K α is found from img class="tex" alt="\operatorname{Pr}(K\leq K_\alpha)=1-\alpha.\," src="http://upload.wikimedia.org/math/b/b/4/bb4772bb6ae01da6b6a3d1d6b3b43097.png" real_src="http://upload.wikimedia.org/math/b/b/4/bb4772bb6ae01da6b6a3d1d6b3b43097.png" title="Kolmogorov–Smirnov test" style="margin:0px;padding:0px;border:0px;list-style:none;" / The asymptotic power of this test is 1. If the form or parameters of F ( x ) are determined from the X i , the inequality may not hold. In this case, Monte Carlo or other methods are required to determine the rejection level α .
Chi-square, Fisher's exact, and McNemar's test(转自 http://yatani.jp/HCIstats/ChiSquare ) Table Of Contents Chi-square test Effect size R code example How to report Fisher's exact test R code example How to report McNemar's test Effect size R code example McNemar's test and binomial test How to report Chi-square test A Chi-square test is a common test for nominal (categorical) data. One application of a Chi-square test is a test for independence. In this case, the null hypothesis is that the occurrence of the outcomes for the two groups is equal. For example, you have two user groups ( e.g. , male and female, or young and elderly). And you have nominal data for each group, for example, whether they use mobile devices or which OS they use. So, your data look like this. If your data of the two groups came from the same participants ( i.e. , the data were paired), you should use McNemar's test . Own device A Don't own device A Male 25 5 Female 15 15 Windows Mac Linux Young 16 11 3 Old 21 8 1 And now you are interested in figuring out whether the outcomes for the two groups were statistically equal. The assumption of Chi-square is that the samples are taken independently or are unpaired . If not, you need to use McNemar's test. And if you have only a small sample size, you should use Fisher's exact test . Effect size The effect size of a Chi-square test can be described by phi or Cramer's V . If your data table is 2 x 2, you will calculate phi ( k =2 in the equation below) and otherwise, Cramer's V ( k 2 in the equation below) . But the calculation is pretty much the same and it is as follows: , where N is the total number of the samples, and k is the number of the rows or columns, whichever smaller, in your data table. And the chi-squared here is the value without any correction. Here are values which are considered small, medium and large sizes. small size medium size large size Cramer's phi or V 0.10 0.30 0.50 R code example Let's use the examples above. First, prepare the data. data - matrix(c(25, 5, 15, 15), ncol=2, byrow=T) data2 - matrix(c(16, 11, 3, 21, 8, 1), ncol=2, byrow=T) And run a Chi-squared test. chisq.test(data) Pearson's Chi-squared test with Yates' continuity correction data: data X-squared = 6.075, df = 1, p-value = 0.01371 chisq.test(data2) Pearson's Chi-squared test data: data2 X-squared = 2.1494, df = 2, p-value = 0.3414 So, the first example has a significant difference, which means the ownership of device A significantly differs between male and female users. The effect size of the first test can be calculated with vcd package: library(vcd) assocstats(data) X^2 df P( X^2) Likelihood Ratio 7.7592 1 0.0053440 Pearson 7.5000 1 0.0061699 Phi-Coefficient : 0.354 Contingency Coeff.: 0.333 Cramer's V : 0.354 For a 2x2 table, you can also calculate the odds ratio . The odds ratio is how the probability of the phenomena is affected by the dependent variable. This can be calculated as ad / bc . Own device A Don't own device A Male a = 25 b =5 Female c = 15 d = 15 (25 * 15) / (5 * 15) 5 How to report You can report the results of a Chi-square test like this: Our Chi-square test with Yates' continuity correction revealed that the percentage of the ownership of device A significantly differed by gender ( (1, N = 60) = 6.08, p 0.01, = 0.35, the odds ratio is 5.0) . Fisher's exact test You can instead use Fisher's exact test if your sample size is small. It is hard to say how many samples are small, but in general, it is better to use a Fisher's exact test than a Chi-square test when you have small than 10 in any cell of your data table (like the examples above). R code example Running a Fisher's exact test is pretty similar to Chi-square. fisher.test(data) Fisher's Exact Test for Count Data data: data p-value = 0.0127 alternative hypothesis: true odds ratio is not equal to 1 95 percent confidence interval: 1.335859 20.757326 sample estimates: odds ratio 4.859427 How to report How to report the results of a Fisher's exact test is pretty much the same as a way to report the result of Chi-square test . Unlike Chi-square test, you don't have any statistics like chi-squared. So, you just need to report the p value. Some people include the odd ratio with the confidence intervals. McNemar's test McNemar's test is basically a paired version of Chi-square test. Let's say you asked whether the participants liked the device before and after the experiment. After experiment Yes No Before experiment Yes 6 2 No 8 4 Here, what you want to test is whether the number of the participants who liked the device were significantly changed between before and after the experiment. Effect size The effect size of an Fisher's exact test can be calculated in the same way as a Chi-square test . R code example Running a McNemar's exact test is pretty similar to Chi-square. data - matrix(c(6, 2, 8, 4), ncol=2, byrow=T) mcnemar.test(data) McNemar's Chi-squared test with continuity correction data: data McNemar's chi-squared = 2.5, df = 1, p-value = 0.1138 Thus, we cannot reject the null hypothesis, and it means that the number of the participants who liked the device were not significantly changed between before and after the experiment. As you can see here, mcnemar.test() automatically makes correction for continuity. You can disable it with correct=F option, and the results will become the same with the function for Cochran's Q test . McNemar's test and binomial test In SPSS, the binomial distribution is used for McNemar's test. Thus, the results look different from those you can get in R. A binomial test is very similar to McNemar's test, but its null hypothesis is that the ratio of the two categories is equal to an expected distribution. In most cases, a binomial test is used for testing whether two categories are equally likely to occur. Question 2 Yes No Question 1 Yes a b No c d More precisely, you need to use a binomial test rather than McNemar's test if b+ c in the 2x2 table is small. However, in R, you can run McNemar's test with continuity correction, so it will cause a big problem because the results of a binmoal test and McNemar's test with continuity correction become similar. If you want to do a binomial test like SPSS does, you need to use binomial function. And you need two numbers, which is the total count for the cases where the participants flipped the responses ( i.e. , b+ c . In the example we are using, 2 + 8 = 10), and the number of one of these two cases ( i.e. , 2 or 8). After experiment Yes No Before experiment Yes 6 2 No 8 4 binom.test(2, 10, 0.5) Exact binomial test data: 2 and 10 number of successes = 2, number of trials = 10, p-value = 0.1094 alternative hypothesis: true probability of success is not equal to 0.5 95 percent confidence interval: 0.02521073 0.55609546 sample estimates: probability of success 0.2 In this case, the p value is pretty close regardless of the ways to do a McNemar's test. How to report How to report the results of a McNemar's test is pretty much the same as those of a Chi-square test. See here for more details.
在生物学特别是基因组学的研究工作中,经常会遇到多重假设检验(multiple testing)的问题;此时,得到的原始p值需要进行校正后才能使用,那么哪种校正方法更加适合自己的研究工作呢?p-values, false discovery rates(FDR) 和 q-values有什么不同?它们分别代表什么意义?对于统计科班的同学来说,这不过是小菜一碟;但对于纯生物出身的同学来说,别说去看公式了,光是听听就觉得头大!不过幸运的是,有牛人(William S Noble)了解我们的苦衷,于是一篇nature biotechnology的文章诞生了——《How does multiple testing correction work?》。这片文章不长,只有3页,用不了多长时间就可以看完。更加令人高兴的是,全篇没有一个让人头大的公式;了解基本的统计学知识、特别是p值的相关概念之后,阅读这片文章就不会有太大的困难了。作者以一个生物学例子贯穿全篇,这个例子对于大多数生物专业的同学来说都非常容易理解——在人的21号染色体上寻找CTCF(一个高度保守的锌指DNA结合蛋白)的潜在结合位点。作者先介绍了零假设(null hypothesis),进而引出了p-value的概念。之后,解释了为什么原始p值不能够直接使用,从而过渡到p值校正的话题。在这一部分,作者层层深入,以简洁明了的语言介绍、解释了Bonferroni adjustment、false discovery rate (FDR)、q-value和local FDR的概念、由来、意义等基本但非常重要的知识。最后作者给出了实际应用时的指导建议,并以点睛之笔概括总结了全文中的要点。如果你的工作涉及p值的校正、FDR、q值等概念,这篇文章绝对胜任引你入门的角色(但绝不仅限于此!)。 文章链接: http://www.seq.cn/forum.php?mod=viewthreadtid=3504 1 2 3 When prioritizing hits from a high-throughput experiment, it is important to correct for random events that falsely appear significant. How is this done and what methods should be used? Imagine that you have just invested a substantial amount of time and money in a shotgun proteomics experiment designed to identify proteins involved in a particular biological process. The experiment successfully identifies most of the proteins that you already know to be involved in the process and implicates a few more.
下面对推论统计部分做个系统性的总结。 1 、显著性 显著性的含义是指两个群体的态度之间的人和差异是由于系统因素而不是偶然性因素的影响。 显著水平值得是你愿意承担的风险水平或者概论水平。显著水平是不能 100% 确信试验中观察到的结果是由于处理因素或需要检验的因素引起的。一般我们认为在显著水平在 0.05 和 0.01 之间时是可接受的。如果显著水平是 0.01 ,意味着在任何一个零假设检验中,只有 1% 的可能性是零假设为真,而你拒绝了零假设,并且在群体之间实际上根本没有差异的情况下得出群体之间有差异的结论。即只有 1% 的可能性会出现下面的情况:零假设为真,但是你却认为零假设为假。 统计显著性是指零假设为真的情况下拒绝零假设所要承担的风险水平。可以理解为,对零假设判断错误的可能性或者概论。 2 、不同分析目标对应不同的分析方法 针对不同的分析人群和目标,需要选择合适的检验分析方法。主要方法选择可以归结为如下树状结构图。每次选择方法时,只需要按照对应的情况进行选择即可。(详细过程见附件) 2.1 两个群体的 t 检验 当你需要进行两个群体在一个或者多个变量上的差异时,需要对这两个群体进行 t 检验分析。 T 检验的一个主要的假设时两个群体中每个群体的变异性的量时相等的。这是方差齐性假定。这种假定很少被破坏,虽然这种可能确实存在。 2.1.1 独立样本的 t 检验 这里的独立性可以理解为两个群体在任何方面都不相关。独立样本的 t 检验,指的是针对两个独立群体进行一次测试,研究中的每个参与者只接受一次测试。 步骤: ( 1 )零假设和研究假设的表述 零假设: H 0 : μ 1 = μ 2 研究假设: H 1 : X 1 ≠ X 2 ( 2 )设置零假设的风险水平(或显著性水平)。一般设置为 0.05 或者 0.01. 这完全由你决定。 ( 3 )独立样本的 t 检验公式 (公式见附件) 其中, X 1 和 X 2 表示群体 1 和群体 2 的均值; n 1 和 n 2 表示群体 1 和群体 2 的参与者数量; s 1 2 和 s 2 2 表示群体 1 和群体 2 的方差。 将具体数值带入公式,计算得出 t 值。这一步可以依靠统计工具如 SPSS 来完成。 ( 4 )使用特定的统计量的临界值分布表确定拒绝零假设需要的值。 这一步需要查表。这里有个名词需要解释: 自由度:对于两个独立群体的均值 t 检验分析,自由度 df= n 1 + n 2 -2 . 不同统计检验,对应的自由度的计算方法可能不同。 ( 5 )比较实际值和临界值 如果实际值大于临界值,就不能接受零假设。如果实际值没有超过临界值,则接受零假设。 ( 6 )结果的解释 假设最后结果如下: t 58 =-0.18 , p 0.05. T 表示我们所用的检验统计量; 58 是自由度数值; -0.18 是实际值,是使用公式计算得到的; p 0.05 表示对零假设的任何检验来说,两个群体的差异是由于随机因素的可能性大于 5% ,结果是非显著的。即接受了零假设,两个群体之间的差异可以认为是由于群体间的随机因素引起的,不是由于某种特定因素引起的。 2.1.2 非独立样本的 t 检验 当需要对测试人群在两种不同的条件下进行相同的测试研究时,就需要用非独立样本的 t 检验分析来检验两个群体之间的差异性。注意,这里必须是两个相同人群,在不同条件下进行两次相同的测试。比如针对群体 1 和群体 2 ,在对这两个群体进行阅读培训之前进行一次阅读能力测试,培训结束后进行一次相同的阅读能力测试。 检验过程与独立样本的 t 检验过程一样,只是公式略有不同。非独立样本的 t 检验公式为: (公式见附件) 其中, D 表示两组数据间差异的总和 ; D 2 表示两组数据间差异的平方和; 对于结果的解释: 例如 t 24 =2.45 , p 0.05 24 表示自由度数值; 2.45 是实际值,是通过上面的公式计算得到的; P 0.0 5 表示对零假设的任何检验来说,群体间的差异是由于随机因素影响的概率小于 5% 。也就是说有其他特定因素影响导致了群体间的差异。即拒绝了零假设,存在显著性差异。 2.2 两个以上群体的方差分析—— F 检验 任何分析如果只有一个维度或者一个处理变量,分组因素有两个以上的层级,而且关注不同的群体在平均成绩上的差异,就需要采用方差分析,也就是 f 检验。简单方差分析值分析一个因素或者一个处理变量(如群体身份),而有两个以上的群体收到这个因素的影响。当需要分析多个因素或者处理变量时,就需要用到析因分析。 方差分析公式: F= MS between/ MS within 其中, MS between 是组间差异平方和的平均值, MS between = df between / ( X 2 n – X 2 N ) ; df between = k-1; K 是群体的数量; df within =N-k ; N 是总的样本规模; MS within 是组内差异平方和的平均值, MS within = df within / ( X 2 - X 2 n ) ; 从上面的过程可以看出,计算一次 F 值非常麻烦, SPSS 可以完成这个计算过程,直接得到 F 值。 方差分析的整体过程与独立样本的 t 检验分析的过程类似,只是具体的临界值和计算公式不一样。这里不再赘述。 2.3 析因分析 析因分析是对简单方差分析的一种进一步的深入和补充。简单方差分析只能对一种因子或者维度进行分析,但是析因分析可以加入两个或以上,分别对每个因子的影响和因子相互作用产生的影响逐一进行分析,也就是研究者可以分析每一个因素的效应,同时可以通过交互效应分析两者共同的效应。 2.3.1 析因方差分析中的主效应 方差分析的最初目标是检验两个或两个以上群体之间的差异。如果数据分析表明某个因素的不同层级之间存在差异,我们认为存在主效应。 2.3.2 析因方差分析中的交互效应 如果数据分析表明,因素之间相互影响产生一定结果和影响时,我们认为存在交互效应。比如分析男性和女性分别在高强度和低强度训练下体重减轻程度时,不管是男性还是女性或者是在高强度或者低强度下都不重要,但是同时处于两种条件下就很重要,高强度项目中女性减少体重比男性减少的体重多,而在低强度项目中男性减少的体重比女性减少的体重多。这就是性别和项目强度交互影响产生的交互效应。 由于析因分析计算太过复杂,因此一般情况下不会手动计算,直接依靠统计工具得到最后结果。 注意,主效应和交互效应并不是每次都会存在,有可能在某次分析中不存在主效应,而存在交互效应。也可能只存在主效应,而不存在交互效应,或者是二者同时存在。 2.4 使用相关系数检验关系 当需要检验两个变量之间的关系时,需要使用相关系数显著性的 t 检验。相关系数检验只检验变量之间的关系而不是群体之间的差异。 自由度 df = n-2 ; 由于一些统计学家已经计算了在不同显著水平下( 0.01 和 0.05 )不同样本规模的单侧检验和双侧检验临界 r 值,或者可以借助统计软件完成这个步骤,因此不在详述计算过程。 2.5 线性回归 可以通过线性回归来进行估计。估计就是使用已经收集的数据集计算变量如何相关,然后使用相关系数以及 X 的信息来估计 Y 。其实就是建立 X 和 Y 的函数关系,然后通过带入已知的 X 值,计算 Y 的近似估计值 Y’ 。 方程为 Y’ = bX + a ; Y’ 表示一直 X 值的 Y 的估计值; b 表示直线的斜率或者方向 ;b = XY –( X Y/n ) X 2 - ; a 表示直线与 y 轴相交的点。 a = Y -b X n ; 回归线又称为最优拟合线,并不是所有回归线都是直线,可能是曲线。 既然回归线是一条评估的拟合线,那么我们需要使用一定的方法来评估我们的估计有多么精确。如果我们考虑所有的偏差,计算每个数据点偏离与估计的数据点的平均数量,即标准估计误差。这个值告诉我们估计的不准确性程度。 线性回归可以对多个变量进行估计,即多元估计变量。那么需要遵守一个原则,即需要选择的独立变量 X 与被估计变量 Y 相关,并且尽量选择相互独立或者不相关的变量,但是都要跟 Y 相关。 哎,科学网的博客图片没办法直接复制,直接加附件上传吧。上面的公式都被错位了,大家如果需要还是直接下载附件哈。
R 语言检验两个向量是否完全一致 熊荣川 六盘水师范学院生物信息学实验室 xiongrongchuan@126.com http://blog.sciencenet.cn/u/Bearjazz 在更新数据时可能会因为一些小小的误操作而导致,操作前后的两组数据有细微的差别,乍一看不能容易发现,使用 R 语言两向量的一致性判别,可以精确的进行判别和纠错,下面是代码过程及相应的结果和备注。 rm(list = ls()) # 清空控制台所有向量 a=c("A","T","G","C") # 定义向量 a b=c("A","T","G","C") # 定义向量 b all(a == b) # 检验 a 和 b 是否完全一致 TRUE a=c("A","T","G","C") # 定义向量 a b=c("A","C","G","T") # 定义向量 b all(a == b) # 检验 a 和 b 是否完全一致 FALSE a == b # 查看详细检验 TRUE FALSE TRUE FALSE ind = c(a == b) a # 显示 a 中和 b 不匹配的元素 "T" "C" b # 显示 b 中和 a 不匹配的元素 "C" "T" 就这么简单,祝您科研愉快!
-- 引载自Chris同学的读书笔记,3Q! (六)检验分子水平自然选择的方法 在选择主义与中性主义的争论中,中性理论提出了很多的假设,其中的许多涉及到群体内等位基因频率分布,以及种内 - 种间遗传变异的关系。因此,可以利用统计学模型来验证中性学说的正确性,即把中性理论作为统计学检验的零假设( null hypothesis ),非中性选择作为选择性假设( alternative hypothesis ),如果这个零假设被显著地拒绝( significantly rejected ),那么中性假设将被认为是不合适的( Kimura and Ohta 1971 )。 关于在分子水平验证选择的方法, Garrigan 和 Hedrick ( 2003 )认为可以按照种群的当前世代,种群的短期历史和物种的长期演化历史三种时间尺度来划分为三类。然而,选择是一个长期作用的过程,种群的当前世代体现出来的临时状态无法真实反映选择的作用;并且这种时间尺度的划分也不利于寻找种内 - 种间遗传变异所反映的选择信号。 Nielsen ( 2005 )则把选择检验分为群体遗传学检验( population genetic approaches )和比较数据检验( comparative data approaches )。 Biswas 和 Akey ( 2006 )从基因组学的角度出发,将选择检验的方法分为种内多态性,种内多态性与种间分歧,和种间检验三类。事实上,不论如何划分,不同的检验方法都有不同的数据类型作为检验对象。因此,在这篇综述里我将按照数据类型的不同对目前常用的统计检验方法进行整理和归纳。 ( 1 )基于群体内等位基因频率分布的中性检验 在核酸的碱基测序时代之前,群体遗传多样性的研究手段主要是对遗传标记的电泳图谱进行分析,其中等位基因的杂合度( allele heterozygosity )曾经是一个普遍用于描述遗传多样性的指标。以某单一等位基因位点为例,在一个个体数为 1000 的群体里,如果其中 50 个个体在该位点是杂合子,那么我们可以简单地把( Ho ) =50/1000=0.05 作为该位点的表观杂合度;说明该种群在以这个位点为遗传标记时得到的遗传多样性程度不高,即仍有 95% 的个体是纯合子。这种评估方式适用于小片段的蛋白质或核酸序列(如几十或者几百个氨基酸或碱基),但不适用于较长片段的研究。事实上,在自然状态下,核酸水平上的变异是比较丰富的,尤其从大片段的尺度来看。例如比较两条长度为 10,000 bp 的等位基因,如此长度的序列几乎可以肯定他们是杂合的,因为序列越长,里面的变异越丰富,那么可以想象该位点在群体里杂合度 Ho 接近 1 。因此,在对核酸序列进行群体遗传多样性分析时,考虑两条序列间存在多少差异所获得的遗传多样性信息要远远大于判断他们是纯合子还是杂合子( Li 1997 )。 在后来发展起来的群体遗传学研究中,有三个重要指标被运用于评估核酸遗传多样性( Nei 1987; Li 1997 )。第一个是 ∏ ,即将所研究群体的所有核酸序列中任意两条不同序列的碱基差异数取平均值;这个指标对等位基因频率依赖很大。第二个是 K ,即分离位点数( number of segregating sites ),现在也被称为 SNP ( single nucleotide polymorphism ),是指所有序列排列比对后存在变异的碱基位点数目;这个指标依赖于等位基因数目而与等位基因频率无关。第三个是 Na ,即等位基因数( number of alleles )。此外,有一个非常关键的反映种群动态的参数 θ 将以上三个指标在数学上联系起来;这里 θ=4 N e μ ,其中 N e 为有效种群大小, μ 为每一代的序列突变率( Watterson 1975; Tajima 1983 )。有两种公认的 θ 估值,一个是 Watterson 估值( Watterson’s estimator, θ W ),把 θ 与 K 联系起来,即 θ W =K/a ,其中 a= ( Watterson 1975 );另一个是 Tajima 估值( Tajima’s estimator, θ T ),即 θ T =∏ ( Tajima 1983 )。从理论上说,在中性条件下,应当有 θ T =θ W =4 N e μ 的平衡状态。因此, Tajima ( 1989 )设计了 D 值检验( Tajima’s D ),即 D= ,通过统计学模型来验证中性突变假说。 Tajima’s D 值检验的作用原理是( Tajima 1989 ):在原有的平衡状态中( θ T =θ W =4 N e μ ),所以 D=0 。但是,如果群体中存在许多低频率的等位基因(稀有等位基因),可以期望 K/a 不断增大而 ∏ 并未受到严重影响,因为后者主要是由高频率等位基因决定的。于是有 θ T θ W ,则 D0 。相反,当群体中是中等频率的等位基因占主导时,可以期望 ∏ 增大而 K/a 不受影响;这时 θ T θ W , D0 。 Tajima ( 1989 )把过多低频率等位基因的存在归咎为定向选择时,选择性清除下选择性清除会削弱原有等位基因的在群体中的频率,而使新等位基因以低频率补充进来成为稀有等位基因。相反,如果是中等频率的等位基因占主导,则可能是平衡选择的结果,或者是种群大小在经历瓶颈时使稀有等位基因丢失。因此,当 Tajima’s D 显著大于 0 时,可用于推断瓶颈效应和平衡选择;当 Tajima’s D 显著小于 0 时,可用于推断群体规模放大和定向选择。由于平衡选择与定向选择都属于正选择的范畴,因此,只要 D 值显著背离 0 ,就可能是自然选择的结果;而当 D 值不显著背离 0 时,则中性零假说则不能被排除。 之后, Fu 和 Li ( 1993 )提出了与 Tajima’s D 略为不同的方法来检验中性进化,即 Fu and Li’s D F test 。他们考虑的是可以获得外类群的情况,因而对一组给定的等位基因序列可以构建一颗有根树。在这棵树上,总突变数为 y ,内部分枝突变数为 y i ,外部分枝的突变数目为 y e ,则 y=y i +y e 。这里 y 和 y e 的数学期望值分别为 E(y)=a*θ , E(y e )= θ ,其中 a= 。如果发生了选择作用,那么外部分枝突变数将会偏离期望值,而内部分枝突变数并未受到严重影响。因此,可根据与 Tajima’s D 类似的策略,构建统计模型来验证中性零假说。此外, Fay 和 Wu ( 2000 )构建了 H 检验( Fay and Wu’s H test ),用以测试高频率变异与中等频率变异的差异。他们认为在中性占主流的状态下,并不期望会出现很多高频率的变异,因而仅仅根据少数存在的高频率的变异就可以推断 “ 搭车效应 ” 。在果蝇的一些低频重组的区域中, H 检验观察到了许多高频率变异,因此, Fay 和 Wu ( 2000 )推断果蝇中的这些高频变异可能是由于 “ 搭车效应 ” 时正选择保留了有利变异并使其以高频率在群体中存在。 到目前为止, Tajima’s D , Fu and Li’s D F test 和 Fay and Wu’s H test ,可能是针对群体内的等位基因频率被运用得最广泛的中性检验模型( Nielsen 2005 )。 原文来自: http://hi.baidu.com/wangjuan730/blog/item/02724f3bfb48f92d70cf6cd8.html
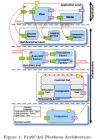
Reconfigurable Run-Time Support for Distributed Service Component Architectures Rémi Mélisson, Philippe Merle, Daniel Romero, Romain Rouvoy, and Lionel Seinturier ASE’10, September 20–24, 2010, Antwerp, Belgium ABSTRACT SCA (Service Component Architecture) is an OASIS standard for describing service-oriented middleware architectures. In particular, SCA promotes a disciplined way for designing distributed architectures based on a component model and an Architecture Description Language (ADL). However, SCA does not cover the deployment and the run-time management of SCA applications. In this paper, we therefore describe the FraSCAti platform, which provides run-time support, deployment capabilities, and run-time management for SCA. Compared to state-of-the-art platforms, FraSCAti brings a dynamic reflective support to SCA and enables both introspecting and reconfiguring service-oriented architectures at run-time. To achieve this capability, the components are completed by a dedicated container, which is automatically generated by the platform. Furthermore, FraSCAti is a highly configurable platform that can be easily customized by finely selecting the features and functionalities which need to be included. In this way, the platform can be adapted to different application needs and middleware environments. Keywords: Component-Based Software Engineering (CBSE), Middleware, Service Component Architecture (SCA), Service-Oriented Architecture (SOA) 1. INTRODUCTION SCA 2. THE FRASCATI PLATFORM FraSCAti:a reflective platform for deploying, hosting, and managing SCA applications, and its different subsystems are implemented as SCA components. the architecture of the platform (illustrated in Figure 1) relies on the four following layers: -- Kernel Level. -- Personality Level -- Run-time Level. -- Non-Functional Level. 3. RELATED WORK -- OpenCOM -- Hadas -- K-Component Reconfigurable run-time support for distributed service component architectures.pdf I comment: whether or SCA is the component-based model for SOA?
-- 引载自Chris同学的读书笔记,3Q! (六)检验分子水平自然选择的方法 在选择主义与中性主义的争论中,中性理论提出了很多的假设,其中的许多涉及到群体内等位基因频率分布,以及种内 - 种间遗传变异的关系。因此,可以利用统计学模型来验证中性学说的正确性,即把中性理论作为统计学检验的零假设( null hypothesis ),非中性选择作为选择性假设( alternative hypothesis ),如果这个零假设被显著地拒绝( significantly rejected ),那么中性假设将被认为是不合适的( Kimura and Ohta 1971 )。 关于在分子水平验证选择的方法, Garrigan 和 Hedrick ( 2003 )认为可以按照种群的当前世代,种群的短期历史和物种的长期演化历史三种时间尺度来划分为三类。然而,选择是一个长期作用的过程,种群的当前世代体现出来的临时状态无法真实反映选择的作用;并且这种时间尺度的划分也不利于寻找种内 - 种间遗传变异所反映的选择信号。 Nielsen ( 2005 )则把选择检验分为群体遗传学检验( population genetic approaches )和比较数据检验( comparative data approaches )。 Biswas 和 Akey ( 2006 )从基因组学的角度出发,将选择检验的方法分为种内多态性与种间分歧,和种间检验三类。事实上,不论如何划分,不同的检验方法都有不同的数据类型作为检验对象。因此,在这篇综述里我将按照数据类型的不同对目前常用的统计检验方法进行整理和归纳。 ( 1 )基于群体内等位基因频率分布的中性检验 在核酸的碱基测序时代之前,群体遗传多样性的研究手段主要是对遗传标记的电泳图谱进行分析,其中等位基因的杂合度( allele heterozygosity )曾经是一个普遍用于描述遗传多样性的指标。以某单一等位基因位点为例,在一个个体数为 1000 的群体里,如果其中 50 个个体在该位点是杂合子,那么我们可以简单地把( Ho ) =50/1000=0.05 作为该位点的表观杂合度;说明该种群在以这个位点为遗传标记时得到的遗传多样性程度不高,即仍有 95% 的个体是纯合子。这种评估方式适用于小片段的蛋白质或核酸序列(如几十或者几百个氨基酸或碱基),但不适用于较长片段的研究。事实上,在自然状态下,核酸水平上的变异是比较丰富的,尤其从大片段的尺度来看。例如比较两条长度为 10,000 bp 的等位基因,如此长度的序列几乎可以肯定他们是杂合的,因为序列越长,里面的变异越丰富,那么可以想象该位点在群体里杂合度 Ho 接近 1 。因此,在对核酸序列进行群体遗传多样性分析时,考虑两条序列间存在多少差异所获得的遗传多样性信息要远远大于判断他们是纯合子还是杂合子( Li 1997 )。 在后来发展起来的群体遗传学研究中,有三个重要指标被运用于评估核酸遗传多样性( Nei 1987; Li 1997 )。第一个是 ∏ ,即将所研究群体的所有核酸序列中任意两条不同序列的碱基差异数取平均值;这个指标对等位基因频率依赖很大。第二个是 K ,即分离位点数( number of segregating sites ),现在也被称为 SNP ( single nucleotide polymorphism ),是指所有序列排列比对后存在变异的碱基位点数目;这个指标依赖于等位基因数目而与等位基因频率无关。第三个是 Na ,即等位基因数( number of alleles )。此外,有一个非常关键的反映种群动态的参数 θ 将以上三个指标在数学上联系起来;这里 θ=4N e μ ,其中 N e 为有效种群大小, μ 为每一代的序列突变率( Watterson 1975; Tajima 1983 )。有两种公认的 θ 估值,一个是 Watterson 估值( Watterson’s estimator, θ W ),把 θ 与 K 联系起来,即 θ W =K/a ,其中 a= ( Watterson 1975 );另一个是 Tajima 估值( Tajima’s estimator, θ T ),即 θ T =∏ ( Tajima 1983 )。从理论上说,在中性条件下,应当有 θ T =θ W =4N e μ 的平衡状态。因此, Tajima ( 1989 )设计了 D 值检验( Tajima’s D ),即 D= ,通过统计学模型来验证中性突变假说。 Tajima’s D 值检验的作用原理是( Tajima 1989 ):在原有的平衡状态中( θ T =θ W =4N e μ ),所以 D=0 。但是,如果群体中存在许多低频率的等位基因(稀有等位基因),可以期望 K/a 不断增大而 ∏ 并未受到严重影响,因为后者主要是由高频率等位基因决定的。于是有 θ T θ W ,则 D0 。相反,当群体中是中等频率的等位基因占主导时,可以期望 ∏ 增大而 K/a 不受影响;这时 θ T θ W , D0 。 Tajima ( 1989 )把过多低频率等位基因的存在归咎为定向选择时,选择性清除会削弱原有等位基因的在群体中的频率,而使新等位基因以低频率补充进来成为稀有等位基因。相反,如果是中等频率的等位基因占主导,则可能是平衡选择的结果,或者是种群大小在经历瓶颈时使稀有等位基因丢失。因此,当 Tajima’s D 显著大于 0 时,可用于推断瓶颈效应和平衡选择;当 Tajima’s D 显著小于 0 时,可用于推断群体规模放大和定向选择。由于平衡选择与定向选择都属于正选择的范畴,因此,只要 D 值显著背离 0 ,就可能是自然选择的结果;而当 D 值不显著背离 0 时,则中性零假说则不能被排除。 之后, Fu 和 Li ( 1993 )提出了与 Tajima’s D 略为不同的方法来检验中性进化,即 Fu and Li’s D F test 。他们考虑的是可以获得外类群的情况,因而对一组给定的等位基因序列可以构建一颗有根树。在这棵树上,总突变数为 y ,内部分枝突变数为 y i ,外部分枝的突变数目为 y e ,则 y=y i +y e 。这里 y 和 y e 的数学期望值分别为 E(y)=a*θ , E(y e )= θ ,其中 a= 。如果发生了选择作用,那么外部分枝突变数将会偏离期望值,而内部分枝突变数并未受到严重影响。因此,可根据与 Tajima’s D 类似的策略,构建统计模型来验证中性零假说。此外, Fay 和 Wu ( 2000 )构建了 H 检验( Fay and Wu’s H test ),用以测试高频率变异与中等频率变异的差异。他们认为在中性占主流的状态下,并不期望会出现很多高频率的变异,因而仅仅根据少数存在的高频率的变异就可以推断 “ 搭车效应 ” 。在果蝇的一些低频重组的区域中, H 检验观察到了许多高频率变异,因此, Fay 和 Wu ( 2000 )推断果蝇中的这些高频变异可能是由于 “ 搭车效应 ” 时正选择保留了有利变异并使其以高频率在群体中存在。 到目前为止, Tajima’s D , Fu and Li’s D F test 和 Fay and Wu’s H test ,可能是针对群体内的等位基因频率被运用得最广泛的中性检验模型( Nielsen 2005 )。 D 0 suggests either a recent population bottleneck or some form of balancing selection. D 0 suggests either population expansion or purifying selection. A quick check in Web of Science reveals that the paper in which Tajima described this approach has been cited over 3100 times since 1994|900 times since I last taught this course two years ago. Clearly it has been widely used for interpreting patterns of nucleotide sequence variation. Although it is a very useful statistic, Zeng et al. point out that there are important aspects of the data that Tajima's D does not consider. As a result, it may be less powerful, i.e., less able to detect departures from neutrality, than some alternatives. 到目前为止, Tajima’s D , Fu and Li’s D F test 和 Fay and Wu’s H test ,可能是针对群体内的等位基因频率被运用得最广泛的中性检验模型( Nielsen 2005 )。 Tajima Test(Tajima’s D)是由日本学者Fumio Tajima在1989年提出的。该检验的目的是鉴定目标DNA序列在进化过程中是否遵循中性进化模型。进行Tajima检验时,要求提供至少由三条同源 序列组成的alignment。Tajima检验将计算并标准化目标序列alignment中每条序列的分离位点数目(number of segregating sites)以及每条序列的核苷酸多样性的值。如果这两个值在统计上被认为差异显著,则拒绝零假设(the null hypothesis),认为目标序列的进化不遵循中性模型,反之,则接受零假设,认为目标序列在进化上遵循中性模型。 实际计算时,Tajima定义了D,作为统计检验量,因此该检验又被称为Tajima’s D。 在实际工作中,可以用软件MEGA 4或DnaSP很方便的进行计算 ( 2 )基于连锁不平衡的中性检验 这里面首先涉及的参数是等位基因频率( allele frequency ),基因型频率( genotype frequency )和单倍型频率( haplotype frequency )。在无视连锁的情况下,最简单的单一位点模型是 “ 哈迪 – 温伯格平衡 ” ( Hardy–Weinberg equilibrium )模式。假设在单一位点上有两种等位基因 A 和 a ,那么该群体存在三种基因型: AA , Aa 和 aa 。如果用 p 表示 A 的等位基因频率, q 表示 a 的等位基因频率,那么在经典的 Mendel 的基因分离定律和独立分配定律下, p 2 为 AA 的基因型频率, 2 pq 为 Aa 的基因型频率, q 2 为 aa 的基因型频率,则有 p 2 +2 pq + q 2 =1 。哈迪 – 温伯格平衡模式认为( Hardy 1908; Weinberg 1908 ),对于一个理想群体,即无穷大的随机交配且没有任何进化压力的群体,基因型频率将以 p 2 , 2 pq 和 q 2 的比例存在于随机交配后的各代中,等位基因频率不会逐代发生改变,故而这个基因座位的基因库不会发生进化。 事实上,当两对性状或者考虑两个等位基因座位时,我们必须考虑有可能的连锁和重组现象。假设研究对象为两个基因座位 A 和 B ,每个座位上的等位基因分别是 A 1 和 A 2 , B 1 和 B 2 ,那么用 x 来表示四种单倍型的频率: A 1 B 1 : x 11 A 1 B 2 : x 12 A 2 B 1 : x 21 A 2 B 2 : x 22 而每一个等位基因的频率表示为: A 1 : p 1 = x 11 + x 12 A 2 : p 2 = x 21 + x 22 B 1 : q 1 = x 11 + x 21 B 2 : q 2 = x 12 + x 22 假设两个座位上的等位基因是自由地独立地分配到后代中去,那么以 A 1 B 1 为例,我们可以期望 x 11 (e)= p 1 q 1 。这时单倍型频率的观察值 x 11 (o) 与期望值 x 11 (e) 之间的差异,就可以用来反映连锁不平衡( linkage disequilibrium, LD ): D= x 11 (o) – p 1 q 1 。 连锁平衡( linkage equilibrium, LE )指的就是这种两个座位上的等位基因是自由地独立地分配到后代中去的现象, A 与 B 的组合是完全随机的,因此有 D=0 ,即 LD 为零的状态。我们其实可以把 LE 看作是双位点版本的 “ 哈迪 – 温伯格平衡 ” ,只不过这里是单倍型频率而不是基因型频率。当 D≠0 时,观察值与期望值不符,我们就说这两个等位基因处于连锁不平衡状态。 事实上,重组能打断连锁关系而使在很多代以后 LD 趋向于 0 。假设 c ( 0c1 )为两位点间的充重组率,则在第二代时 A 1 B 1 的单倍型频率为: x' 11 =(1–c)x 11 +c p 1 q 1 ,也可以写成 x' 11 – p 1 q 1 =(1–c) ( x 11 – p 1 q 1 ) ,即 D 1 =(1–c)D 0 。扩展到第 n 代时,有 D n =(1–c) n D 0 。如果 n 趋向于 +∞ ,则 (1–c) n 趋向于 0 ,这时 D n =0 。如果两位点在物理距离上越接近,连锁越紧密,被重组的可能性就越低,则 D n →0 的速率就越慢。 在前面提到的 “ 搭车效应 ” 中,当一个有利突变开始产生时,它是处于完全 LD 状态的,即可视为与其构成单倍型的所有基因完全连锁,而其他单倍型因为不存在这个突变而被选择性清除所消灭( Ennis 2007 )。因此,搭车效应,正选择,选择性清除,连锁不平衡,基因重组以及群体结构相联系组成了一种情况极为复杂的局面,使得基于 LD 检验统计模型的设计成为一个难度极高的挑战。尽管如此,近几年,已经发展出了一些检验方法用于检测与 LD 相关联的自然选择,包括 LRH test ( Sabeti et al. 2002 ), iHS test ( Voight et al. 2006 ), LDD test ( Wang et al. 2006 )等。然而,这些检验效力如何,还需要更多的研究结果来提供证据。 ( 3 )基于种群分化的检验 Wright ( 1931 )首先给出了群体遗传分化系数 F st 的计算公式,用以评估亚群体的分化程度。 Cavalli-Sforza ( 1966 )认为自然选择可能会对群体亚分化形成贡献,因此首次建议用群体间的分化程度来推断自然选择的作用。随后,基于这一想法,大致有两种类型的方法被用于自然选择的测试。一种是 Lewontin-Krakauer test ( Lewontin and Krakauer 1973 ),其作用原理是:群体间的基因流( gene flow )会使大多数位点形成较为平均的遗传分化程度,除了一些明显的异常值( outliers )。大体上,这些异常值可以反映两个方向上的选择:适应性选择能在某些位点上产生异常的高水平遗传分化,而平衡选择则有可能产生低于平均水平的遗传分化。因此,从异常位点与正常位点的遗传分化程度的比较可以推断自然选择。然而,最初的这种 Lewontin-Krakauer test 被 Nei 和 Maruyama ( 1975 )以及 Robertson ( 1975 )所批评,认为在许多群体模型中,该检验中 F st 的期望方差无效。尽管如此,这种思想近年来有逐渐抬头的势态,尤其在运用到基因组大尺度数据的时候。例如 Akey 等 (2002) 在基因组尺度对人类不同群体的 F st 进行了测算推断其中的选择作用; Beaumont 与他的同事( Beaumont and Nichols 1996; Beaumont and Balding 2004 )则设计了更为复杂的统计学模型来检测群体亚分化水平上的异常基因位点。另一种方法是检测不同位点在不同种群中的杂合度水平。例如 Schlotterer 等( 1997 )和 Schlotterer ( 2002 ) 认为在不同种群中通过比较多位点的杂合度,可以把选择的作用和种群统计学效应区分开来。两个不同群体大小的种群,小种群在基因组上的期望变异水平都应低于 大种群。但是受到选择的单位点的变异程度则可能会比基因组水平的差异更低。因此,把群体结构与多个单位点变异联合考虑,则可能推断出基因组上的哪些区域有 可能受到了选择。以上的基于种群分化的检验尽管还没有得到非常普及地应用,但事实上,最近的一些研究表明( Nielsen 2005 ), “ 选择性清除 ” 会强烈地影响群体的亚分化水平,尤其是当 “ 选择性清除 ” 没有来得及涉及到所有研究群体的时候,这种作用更明显。因此,基于群体分化程度来寻找自然选择的信号,仍然是一种可行的思路。 ( 4 )基于种内多态性和种间分歧度之间数据比较的检验 中性理论预言,在中性状态下,种内多态性( intraspecific polymorphism )与种间分歧度( interspecific divergence )之间呈正相关。基于这种预测,有两种检验方法先后被提出,分别是 Hudson–Kreitman–Aguade (HKA) test 和 McDonald-Kreitman (MK) test 。 HKA 检验认为( Hudson et al. 1987 ),在中性状态下,对于不同的基因或者基因位点而言,即使他们之间的变异程度不同,但他们各自的种内多态性与种间分歧度之间的比率将是相同的。例如,组蛋白基因( histone ) 是一个相当保守的基因,突变率很低,因此组蛋白基因种内多态性程度很低,其种间变异度也很低。而对于某些非编码序列来说,其突变率很高,不论在种内种间都 体现了很高的变异水平。但是,在中性条件下,不论对于组蛋白基因还是非编码序列,他们的种内多态性与种间分歧度之间的比率将是相当的。如果自然选择发生了 作用,那么受选择的基因,其种内多态性与种间分歧度之间的比率将偏离中性状态下的期望值。因此,通过同时比较两个或者多个基因各自的种内多态性与种间分歧 度之比, HKA 检验可以检测到自然选择的作用。 但是, HKA 的一个很大的限制就是所研究的基因或者基因位点之间必须是相互独立的,即不存在连锁关系。因为一些研究发现,连锁位点所受到的选择作用,影响的是种内多态性而不是种间的分歧度水平( Maynard-Smith and Haigh 1974; Birky and Walsh 1988 ),因此,除了选择作用之外,任何非中性的影响(如搭车效应或选择性清除)都会使种内多态性偏离期望值。也就是说, HKA 所检测到的自然选择信号,有可能是所研究位点受到了选择,也有可能是与其连锁的位点受到了选择而通过某些非中性效应使所研究位点多态性发生了变化,尽管所研究位点仍然是遵循中性进化的。 随后发展起来的 MK 检验则避免了多基因或者多位点有可能存在的连锁效应所带来的假阳性信号。 MK 检验的基本原理是( McDonald and Kreitman 1991 ):仅仅考查某一基因分别在种内和种间的非同义( nonsynonymous )与同义突变( synonymous )的比值。具体而言,针对 A 与 B 这两个近缘物种,我们把基因 G 在这两个物种里能发现的所有等位基因进行汇总和排列比对后,关注其中存在变异的核苷酸替换位点而忽略无变异位点,即关注分离位点( segregating sites )或 SNP ( single nucleotide polymorphism )位点。在这些所有的变异位点中,假设某一位点如果在来自物种 A 的所有等位基因里都是碱基 C ,而在来自物种 B 的所有等位基因里都是碱基 T ,那么针对 A 与 B 这两个近缘物种而言,这个变异位点我们定义为被固定的替换位点( fixed substitution site );其余的变异位点称为多态性位点( polymorphic site )。如果用 S f , N f , S p 和 N p 分别表示固定同义位点数,固定非同义位点数,多态同义位点数和多态非同义位点数(其中 S 代表 synonymous , N 代表 nonsynonymous , f 代表 fixed , p 代表 polymorphic ),那么在中性状态下,应当有 N f :S f = N p :S p 。对应这四个参数的 2×2 列联表可以用 χ 2 分布或者 Fisher 精准检验来验证零假设。一个显著高于 N p :S p 的 N f :S f 比率意味着两个物种之间的某些非同义替换是由正选择引起的;如果 N f :S f 显著地低于 N p :S p ,就意味着负选择降低了两个物种间的非同义替换数目。 ( 5 )基于编码序列的比对中非同义与同义突变的比值 Kimura ( 1977 )最先提出,在蛋白质编码基因中,每非同义位点的非同义替换数( d N )小于每同义位点的同义替换数( d S )。 因为中性主义预言,在蛋白质编码区,大多数非同义突变造成的氨基酸变异会破坏蛋白质原有功能,因此发生在蛋白质编码区的大多数非同义突变都被视为有害突变 而在固定过程中被净化选择所消灭;而同义突变由于不造成氨基酸改变,因此被认为是中性或近中性的而被随机遗传漂变所固定。所以,在固定后体现出来的替换数 的差异上,将会有 d N d S 。同理,如果没有任何选择作用,即所有突变都是中性或者近中性的,则会有 d N = d S 。所以,中性主义并没有排斥负选择(净化选择),当 d N ≤ d S 时,可以认为大多数 “ 被固定下来的突变 ” 是中性或近中性的。相反,如果观察到 d N d S ,则认为非同义突变是有利突变而被正选择所固定。因此,考察比值 d N :d S (也写为 K A :K S )是否大于、小于或者等于 1 ,成为了检测编码序列自然选择作用的有力工具。 以 上五类是目前较为常用的在分子水平检测自然选择作用的方法,其统计学原理均以中性假设作为检验的零假设,当中性零假设被显著地拒绝时,则认为检测到了自然 选择的信号。近年也发展了一些新的检验方法,但大多都是基于以上五类方法的改进或修正。当然,过去也曾有一些较为独立的其它方法,但是现在已经用得不多( Kreitman 2000 )。 (七)自然选择检验中方法学的利与弊 “世上有三种谎言:谎言,该死的谎言,及统计学”(There are three kinds of lies: lies, damned lies and statistics.)。相传这句话由Benjamin Disraeli所创并为马克?吐温(Mark Twain)所推广 。在非正式场合,我会和我所认识的从事统计遗传学的老师或者同事们谈起这句话;然而他们中的一些常会回应我说,“统计学从不说谎,可是说谎者运用了统计学”(Statistics never lie but liars use statistics.)。我想,这两句话各有各的道理。从上个世纪三十年代初开始,Fisher,Wright和Haldane三位数量遗传学家开始大量地把统计数学与遗传学联系起来。之后的几十年里,许许多多的统计学模型被大量运用于进化与遗传领域。然而,这些统计推断的设计中存在的问题使得围绕方法学所展开的争论一直无法停止。上文提到的五类检验分子水平自然选择的方法,虽然是目前较为流行的方法,但是当被不谨慎地使用时,仍然会得到许多不合理甚至是与预期相反的结论;其自身存在的问题和所忽略的漏洞也让很多研究者带来了质疑的声音。 Tajima的D值检验(1989)是基于群体内等位基因频率分布的最早的检验方法,迄今仍是最流行的方法之一。Tajima(1989)发现当D值分布趋近β分布的时候,可以把β分布用来近似地计算显著水平。但是,由于β分布只是一个粗略的估值,Fu和Li(1993)及Simonsen等(1995)认为显著水平应当由计算机模拟来评估。然而,在评估这些统计模型的检验效力的时候,Simonsen等(1995)发现Fu和Li(1993)的D F test,以及Simonsen等自己建立的这些统计方法都比Tajima的D值检验要弱。Nielsen(2005)则指出,这类基于群体内等位基因频率分布的方法,对于群体结构和群体动力学极为敏感,故而其检验效力对群体中的人口统计学因素不够“鲁棒”(robust);同时,基于群体亚分化Fst指标的检验方法也受到类似的困扰。Nei和Kumar(2000)则认为编码序列中的密码子使用偏爱(codon usage bias)会影响此类检验,因此应当慎重看待此类检验的结果。 而与连锁不平衡(LD)相关的检验,如前文所描述的,搭车效应,正选择,选择性清除,连锁不平衡,基因重组以及群体结构相联系组成了一种情况极为复杂的局面,使得基于LD检验的统计模型的设计难度极高。并且,正如Nielsen(2005)所担心的那样,基于LD的自然选择检验对于基因重组率特别地敏感。由于目前已经有研究发现重组率在基因组的不同区段差异很大(McVean et al. 2004),甚至在非常近缘的物种间变化也很大(Ptak et al. 2004; Wall et al. 2003)。因此,不建议在验证分子水平的自然选择时过份关注LD的格局。 HKA 检验的优劣则通常会被拿来与随后发展起来的MK检验作比较。HKA由于比较的是两个或者多个基因的数据,因此能够把由群体动力学效应与选择效应区分开来(Nachman 2006)。例如某个种群经历了严重的群体规模的缩减(群体动力学效应),那么所有考察的基因位点的变异度都会减小,HKA会得到不显著的结果,即不能显著拒绝中性零假说;而当某个位点受到了选择作用,该位点则会体现出与其他位点不一样的变异程度,HKA能得到显著结果。但是,如前文所述,如果所检视的这些基因和位点如果存在某种程度的连锁效应,那么即使检测到选择信号,也不能说明所研究的位点受到了选择。而MK检验由于只针对单一位点,所以如果检测到选择信号,则该位点即为受选择作用的目标。 由于MK检验所体现出来的优势,近年在涉及群体检验时该检验被广泛应用。最初,McDonald和Kreitman(1991)设计该检验并用于检测三种果蝇的乙醇脱氢酶(Adh)基因编码区,得到Nf:Sf =0.412,Np:Sp =0.047,Fisher精准检验得到Nf:Sf显著大于Np:Sp(P=0.006),因此表明果蝇中的非同义替换受到了正选择的作用。随后,Eanes等(1993, 1996)在果蝇的G6pd中也得到了类似的结果。更为令人吃惊的是,近年的一系列利用MK检验来调查黑腹果蝇与其他果蝇物种之间遗传变异的研究,得到的结果都是认为这些果蝇物种间在基因组水平上存在高水平的正选择信号(如,Smith and Eyre-Walker 2002; Sawyer et al. 2007; Shapiro et al. 2007);相反,在人类与黑猩猩之间却很少能检测到正选择的信号(Bustamante et al. 2005; Gojobori et al. 2007)。这种在果蝇中被大量检测到的正选择信号引发了人们的思考:是否MK检验过于宽松而导致许多假阳性信号被检出?因为按照中性主义者历来的看法,自然状态下绝大多数的突变是选择上呈中性或近中性的,故而果蝇基因组中不应该出现如此普遍的正选择信号。由此,关于MK检验效力的质疑声再起。 最初质疑MK检验的声音来自Whittman和Nei(1991),他们认为MK检验没有考虑同一座位的多重替换;如果考虑了这些替换,则McDonald和Kreitman(1991)的数据就不会否定中性理论了。Akashi(1995)则指出如果存在很强的密码子使用偏爱的话,Nf:Sf也会比Np:Sp高得多而无关自然选择。另外在净化选择松缓时(relaxed purifying selection or relaxed selective constraint),即原先很强的负选择压力被减轻时,将等效于受到正选择,也有可能出现Nf:Sf高于Np:Sp的情况(Nei and Kumar 2000)。 事实上,MK检验对净化选择松缓验证的乏力,正好成为了“近中性理论”支持者们的攻击点。Ohta(1993)发现夏威夷果蝇的Nf:Sf要远远高于黑腹果蝇或其他果蝇。之所以选取夏威夷果蝇是因为夏威夷果蝇被认为在物种形成时经历了瓶颈效应(Desalle and Templeton 1988)。按照近中性理论,在经历瓶颈效应时,群体规模变小,因而许多的轻微有害突变可以在小种群里过渡为近中性并为随机遗传漂变所固定。这种多出来的被固定的轻微有害突变使得该群体的Nf增大(这里N代表nonsynonymous,f代表fixed),因为非同义突变往往是有害的。因此,在用MK检验时会得到Nf:Sf大于Np:Sp的结果;这一结果是由于轻微有害突变在瓶颈效应中避开了净化选择(即相当于净化选择松缓),而不是由于受到正选择。所以说MK检验在面临此类问题时会获得正选择的假阳性信号。运用轻微有害突变的近中性理论,还可以较为合理地解释以上提到的果蝇不同种之间与人类–黑猩猩之间MK检验表现的差异(Hughes 2007a)。例如,Bustamante等(2005)比较人类-黑猩猩基因组时得到Nf:Sf =0.60,同时人类中群体中Np:Sp =0.91,于是有Nf:Sf Np:Sp;相反,Shapiro等(2007)在黑腹果蝇中得到Nf:Sf =0.37,Np:Sp =0.31,有Nf:Sf Np:Sp。Li和Sadler(1991)曾发现黑腹果蝇中核酸多样性至少是人类中的5倍,进而推断黑腹果蝇长期的有效种群大小远远大于人类。在黑腹果蝇中,在极大的有效种群大小状态下轻微有害突变被净化选择消灭而不是被固定,因此群体内非同义突变的多样性减小,涉及的参数是Np减小,以至于有Shapiro等(2007)的结果。而现代人起源时则经历了长期的瓶颈作用(Harpending et al. 1998),这期间产生了大量的轻微有害突变。这些轻微有害突变在人类历经瓶颈作用过后的种群恢复壮大过程中,以众多低频率的稀有等位基因的形式出现,正经历着逐步被净化选择消灭的过程。然而,人类历经瓶颈作用过后的种群恢复壮大过程需要很长的历史,有效群体规模难以达到如黑腹果蝇那样的程度,因此净化选择对目前人类群体中轻微有害突变的清除作用有效性还不高。这些众多的轻微有害突变使得人类群体中Np较高,故有以上Bustamante等(2005)MK检验的结果。 鉴于MK检验存在的各种问题,Nei和Kumar(2000)认为,MK中比较的是两对比值的差异(Nf/Sf–Np/Sp),而dN:dS是比较两个独立值的差异(dN–dS),很显然两个量值的比率比他们的差异更容易受到不同因素的干扰,因此dN:dS检验看起来应当比MK检验更有力。 尽管如此,dN:dS检验目前也存在着一些棘手的问题。前面提到过,如果没有任何选择作用,即所有突变都是中性或者近中性的,则对于碱基替换的平均速率会有dN = dS;但是大多数造成蛋白质氨基酸变化的非同义突变,会破坏蛋白原有功能,属有害突变,因而会被负选择清除掉,于是在随机漂变固定后体现出来的碱基替换数的差异上,将会有dN dS。如果,我们假设偶尔产生了一两个能够增强蛋白质功能的非同义突变,即称为有利突变,那么他们会被正选择所固定;其余的非同义突变大部分仍是有害的,会被负选择清除掉;那么在固定之后,体现在平均速率上依然会有dN dS。这时,正选择的信号就会被dN dS所埋没。因此,要想获得dN dS的结果,需要大量的正选择作用;说得再极端一些,即所有的非同义突变都被正选择所固定,与此同时所有的同义突变有一部分被随机固定,另一部分被随机丧失掉,这时一定会有dN dS。可见dN:dS在检测自然选择时是一个保守的方法,因为负选择总是会消灭大多数非同义突变使得dN:dS倾向于1而导致发生在少数有利突变位点上的正选择被忽视(Nielsen 2005)。然而,对于这个问题,Nei(2005)则认为保守的方法似乎更好,可以减少假阳性正选择信号或对净化选择松缓的误判。 尽管有很多不同方法曾用于计算dN或dS的值(如Miyata and Yasunaga 1980; Li et al. 1985; Nei and Gojobori 1986; Li 1993),这些传统方法的共同点都是对序列的所有位点求平均值,即dN或dS反映的是整条比对序列的情况,每个具体位点无差异(Suzuki and Gojobori 1999; Nielsen 2001; Yang 2002)。为了能够检测出平均状态下可能存在的为负选择所掩盖的正选择作用位点,有两类“单位点模式”(single site model)被设计出来,也成为了互有争论的两个阵营:一类是以Yoshiyuki Suzuki的算法为代表的基于最大简约法的单位点模式(maximum parsimony method),另一类则是以Ziheng Yang为首的基于贝叶斯方法的似然率测试 。 Suzuki和Gojobori(1999)设计的简约法模型的工作原理是:首先构建一棵合理的分子系统发育树,运用最大简约法(Fitch 1971; Hartigan 1973)推断出这棵树上所有内部节点的祖先序列。然后,计算整棵树所有节点序列每个编码位点(codon site)上的非同义替换总数(CN)和同义替换总数(CS),以及每一编码位点的平均非同义替换数(SN)和同义替换数(SS)。以dN = dS为零假设,并假设CN和CS服从二项式分布且非同义替换和同义替换发生的概率分别为SN /( SN + SS)和SS/( SN + SS)。如果零假设被拒绝(P0.05),则当CN/SN CS / SS时指示正选择;当CN/SN CS / SS时指示负选择。 Ziheng Yang的基于贝叶斯方法的似然率测试模型则赋予每个编码位点一个ω值(ω=dN:dS),不同编码位点上的这些ω值符合一定的概率分布。最初Nielsen和Yang(1998)及Yang等(2000)设计了M0–M13共十四个不同的概率分布模型,但其中M2,M3和M8较其他模型能给出更可靠的结果(目前Yang推荐的是M2和M8)。以M8为例,M8用来与M7配对进行统计检验,其中M7为零假设,M8为选择性假设。在M7中,所有位点的ω值均只归入0ω≤1这一个范畴,并服从β分布;而在M8中,位点被分归入0ω0≤1和ω11两个范畴。5%显著水平的似然率测试(likelihood-ratio test)用于判断M7与M8的取舍,其中M7与M8的log-likelihood (lnL)值之差的两倍值可用χ2检验来判断,而自由度为M7与M8的自由参数之差。当零假设M7被拒绝时(P0.05),则M8成立。M8中每一位点被归入0ω0≤1或是ω11范畴的概率用贝叶斯-经验-贝叶斯算法(Bayes empirical Bayes, BEB)计算;那些被划入ω11范畴,其BEB后验概率达到95%以上的位点,被认为属于正选择位点。除了“单位点模式”外,Ziheng Yang还设计了“分枝模式”和“分枝-位点模式”(Yang 2007)。 以上这两种方法自诞生以来就一直被拿来做可信度和优劣比较,互有褒贬。Yang(2001)认为使用推断的祖先序列以及在计算替换数量及其之间的差异时所基于的假设过于简单,导致简约法模型不那么可靠。Wong等(2004)则认为简约法模型过于保守。相反,Suzuki则反击认为Yang的模型容易得到高水平的正选择假阳性信号(Suzuki and Nei 2001; 2002)并且即便给予一个错误的拓扑结构Yang的模型依然会检测出正选择信号(Suzuki and Nei 2004)。此外,Shriner等(2003)发现即使发生基因内重组,Yang的模型仍会给出正选择假阳性信号。尽管Ziheng Yang及其同事数次对模型进行了更正和修改(Wong et al. 2004; Yang et al. 2005),但上述问题仍然没有得到完全地解决(Kosakovsky Pond and Frost 2005; Massingham and Goldman 2005; Nei 2005)。并且近年来,随着适应性选择进化研究的跟风热炒,这种争议越发升级(Nozawa et al. 2009a, 2009b; Yang et al. 2009)。而对于这两种“单位点模式”的选择检验,不管是采用哪种方法,Hughes(2007a)认为他们生效的前提是基于一棵给定的100%正确的系统发育树,而事实上正选择造成的平行进化或趋同进化,以及高重组率都会使构建一棵完全正确的基因树变得很困难;此外,这些方法宣称不需要前设,即在不知道所测基因或位点的生物学意义时就可以检测正选择,那么得到正选择位点就难以解释究竟是真的受到正选择作用还是因为净化选择松缓产生了假阳性信号。因此,在检验分子水平的自然选择时,不管使用任何统计模型都要慎重,否则将会得到有争议的结果。 -- 《分子水平的自然选择》写作更新日期:2010年5月10日 -- by勤劳勇敢善良的 Chris Waken --------- 参考文献: Akashi H. 1995. Inferring weak selection from patterns of polymorphism and divergence at silent sites in Drosophila DNA. Genetics. 139:1067-1076. Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, Civello D, Adams MD, Cargill M, Clark AG. 2005. Natural selection on protein-coding genes in the human genome. Nature. 437:1153-1157. Desalle R, Templeton AR. 1988. Founder effects and the rate of mitochondrial DNA evolution in Hawaiian Drosophila. Evolution. 42:1076–1084. Eanes WF, Kirchner M, Yoon J. 1993. Evidence for adaptive evolution of the G6pd gene in the Drosophila melanogaster and Drosophila simulans lineages. Proceedings of the National Academy of Sciences, USA. 90:7475-7479. Eanes WF, Kirchner M, Yoon J, Biermann CH, Wang IN, McCartney MA, Verrelli BC. 1996. Historical selection, amino acid polymorphism and lineage-specific divergence at the G6pd locus in Drosophila melanogaster and D. simulans. Genetics. 144:1027-1041. Fitch WM. 1971. Toward defining the course of evolution: minimum change for a specific tree topology. Systematic zoology. 20:406–416. Fu Y-X, Li W-H. 1993. Statistical test of neutrality of mutations. Genetics. 133:693–709. Gojobori J, Tang H, Akey JM, Wu C-I. 2007. Adaptive evolution in humans revealed by the negative correlation between the polymorphism and fixation phases of evolution. Proceedings of the National Academy of Sciences, USA. 104:3907–3912. Harpending HC, Batzer MA, Gurven M, Jorde LB, Rogers AR, Sherry ST. 1998. Genetic traces of ancient demography. Proceedings of the National Academy of Sciences, USA. 95: 1961–1967. Hartigan JA. 1973. Minimum mutation fits to a given tree. Biometrics. 29:53–65. Hughes AL. 2007a. Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity. 99:364-373. Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Molecular Biology and Evolution. 22:1208–1222. Li W-H, Sadler LA. 1991. Low nucleotide diversity in man. Genetics. 129:513–523. Li W-H. 1993. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. Journal of Molecular Evolution. 36:96–99. Li W-H, Wu C-I, Luo C-C. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Molecular Biology and Evolution. 2:150–174. Massingham T, Goldman N. 2005. Detecting amino acid sites under positive selection and purifying selection. Genetics. 169:1753–1762. McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 351:652–654. McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. 2004. The finescale structure of recombination rate variation in the human genome. Science. 304:581–584. Miyata T, Yasunaga T. 1980. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. Journal of Molecular Evolution. 16:23–26. Nachman MW. 2006. Detecting selection at the molecular level. In: Evolutionary Genetics, Concepts and Case Studies, edited by C.W. Fox and J.B. Wolf. Oxford University Press, Oxford. Nei M, Kumar S. 2000. Molecular evolution and phylogenetics. Oxford University Press, Oxford. Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution. 3:418–426. Nei M. 2005. Selectionism and neutralism in molecular evolution. Molecular Biology and Evolution. 22:2318–2342. Nielsen R. 2001. Statistical tests of selective neutrality in the age of genomics. Heredity. 86:641–647. Nielsen R. 2005. Molecular signatures of natural selection. Annual Review in Genetics. 39:197-218. Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 148:929-936. Nozawa, M, Suzuki, Y, and M. Nei. 2009a. Response to Yang et al.: Problems with Bayesian methods of detecting positive selection at the DNA sequence level. Proc. Natl. Acad. Sci. USA 106:e96. Nozawa, M., Suzuki, Y., and M. Nei. 2009b. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc. Natl. Acad. Sci. USA 106(16):6700-6705. Ohta T. 1993. Amino acid substitution at the Adh locus of Drosophila is facilitated by small population size. Proceedings of the National Academy of Sciences, USA. 90:4548–4551. Ptak SE, Roeder AD, Stephens M, Gilad Y, P??bo S, Przeworski M. 2004. Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biology. 2:849–855. Sawyer SA, Parsch J, Zhang Z, Hartl DL. 2007. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proceedings of the National Academy of Sciences, USA. 104:6504–6510. Shapiro JA, Huang W, Zhang C, Hubisz MJ, Lu J, Turissini DA, Fang S, Wang HY, Hudson RR, Nielsen R, Chen Z, Wu CI. 2007. Adaptive evolution in the Drosophila genome. Proceedings of the National Academy of Sciences, USA. 104:2271–2276. Shriner D, Nickle DC, Jensen MA, Mullins JI. 2003. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genetics Research. 81:115–121. Simonsen KL, Churchill GA, Aquadro CF. 1995. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 141:413-429. Smith NG, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature. 415:1022–1024. Suzuki Y, Gojobori T. 1999. A method for detecting positive selection at single amino acid sites. Molecular Biology and Evolution. 16:1315–1328. Suzuki Y, Nei M. 2001. Reliabilities of parsimony-based and likelihoodbased methods for detecting positive selection at single amino acis sites. Molecular Biology and Evolution. 18: 2179–2185. Suzuki Y, Nei M. 2002. Simulation study of the reliability and robustness of the statistical methods for detecting positive selection at single amino acid sites. Molecular Biology and Evolution. 19:1865–1869. Suzuki Y, Nei M. 2004. False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Molecular Biology and Evolution. 21:914–921. Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 123: 585-595. Twain M (Author) / Neider C (Editor). 2000. The autobiography of Mark Twain. Harper Perennial Modern Classics. Wall JD, Frisse LA, Hudson RR, Di Rienzo A. 2003. Comparative linkage-disequilibrium analysis of the beta-globin hotspot in primates. The American Journal of Human Genetics. 73:1330–1340. Whittam TS, Nei M. 1991. Neutral mutation hypothesis test. Nature. 354:114-116. Wong WSW, Yang Z, Goldman N, Nielsen R. 2004. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 168:1041–1051. Yang Z. 2001. Adaptive molecular evolution. In Handbook of Statistical Genetics. Edited by Balding D, Bishop M, Cannings C. New York, Wiley. 327-350. Yang Z. 2002. Inference of selection from multiple species alignments. Current Opinion in Genetics Development. 12:688–694. Yang Z. 2007. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 24:1586-1591. Yang Z, Nielsen R, Goldman N, Pedersen A-MK. 2000. Codonsubstitution models for heterogeneous selection pressure at amino acid sites. Genetics. 155:431-449. Yang Z, Nielsen R, Goldman N. 2009. In defense of statistical methods for detecting positive selection. Proc. Natl. Acad. Sci. USA 106:E95-E95. Yang Z, Wong WSW, Nielsen R. 2005. Bayes empirical Bayes inference of amino acid sites under positive selection. Molecular Biology and Evolution. 22:1107–1118.
Earthquakes Home ? Audience ? For Public ? Earthquakes Recent Earthquakes 10/17/2008 M 5.8, eastern New Guinea region, Papua New Guinea(巴布新几内亚6.951S, 147.246E) 10/16/2008 M 6.7, offshore Chiapas, Mexico 10/12/2008 M 6.2, Chuquisaca, Bolivia 10/11/2008 M 6.1, Virgin Islands region 10/11/2008 M 5.8, Caucasus region, Russia http://www.iris.edu/hq/audience/public/earthquakes GFZ Potsdam - Earthquake Bulletin New search Legend Origin Time UTC Mag Latitude degrees Longitude degrees Depth km AM Region Name 2008-10-18 18:41:32 5.3 6.29 S 104.63 E 60 A Sunda Strait, Indonesia 2008-10-18 10:26:35 5.2 20.11 S 178.28 W 423 A Fiji Islands Region 2008-10-18 09:42:04 4.3 1.91 S 77.18 W 171 M Ecuador 2008-10-18 09:29:06 4.9 39.64 N 73.82 E 51 M Tajikistan-Xinjiang Border Region 2008-10-18 08:09:50 4.7 47.57 N 145.59 E 401 A Sea of Okhotsk 2008-10-18 00:54:43 5.8 6.94 S 147.25 E 99 A Eastern New Guinea Reg., P.N.G. 2008-10-17 18:53:17 4.2 43.39 N 46.33 E 10 M Eastern Caucasus 2008-10-17 18:42:43 4.2 38.75 N 20.84 E 41 A Greece 2008-10-17 04:10:05 4.8 5.75 S 152.47 E 78 M New Britain Region, P.N.G. 2008-10-17 02:53:58 4.1 14.53 N 92.52 W 45 M Near Coast of Chiapas, Mexico 2008-10-16 23:24:44 4.8 3.96 S 150.77 E 62 M New Ireland Region, P.N.G. 2008-10-16 19:41:30 6.6 14.51 N 92.46 W 63 M Near Coast of Chiapas, Mexico 2008-10-16 18:28:55 4.8 1.82 N 127.39 E 115 M Halmahera, Indonesia 2008-10-16 11:49:29 4.9 42.54 N 142.62 E 108 M Hokkaido, Japan Region 2008-10-16 09:46:17 4.8 31.22 N 103.80 E 10 M Sichuan, China 2008-10-16 06:39:41 4.7 37.17 N 57.89 E 10 M Turkmenistan-Iran Border Region 2008-10-16 04:28:49 5.0 3.10 S 81.35 E 10 A South Indian Ocean 2008-10-15 19:29:29 4.2 39.02 N 23.54 E 10 A Aegean Sea 2008-10-15 18:37:19 3.1 51.34 N 16.01 E 5 M Poland 2008-10-15 16:35:54 4.9 6.49 S 129.66 E 186 A Banda Sea 2008-10-15 14:28:21 5.3 15.69 S 166.81 E 66 A Vanuatu Islands 2008-10-15 08:02:01 5.1 2.01 N 97.66 E 46 A Northern Sumatra, Indonesia 2008-10-15 03:33:09 3.8 51.52 N 16.04 E 10 A Poland 2008-10-15 00:17:53 4.9 15.09 S 72.20 W 100 A Southern Peru 2008-10-14 19:51:39 5.0 4.24 N 69.20 E 10 A North Indian Ocean 2008-10-14 19:00:38 4.2 50.08 N 12.81 E 10 A Germany 2008-10-14 16:52:49 5.3 21.21 S 178.44 W 335 A Fiji Islands Region 2008-10-14 15:06:53 4.9 43.44 N 46.31 E 32 M Eastern Caucasus 2008-10-14 10:17:17 3.4 51.25 N 15.86 E 5 M Poland 2008-10-14 05:49:03 2.8 50.20 N 12.47 E 5 M Germany 2008-10-14 04:05:48 3.4 50.17 N 12.40 E 5 M Germany 2008-10-14 04:01:35 3.7 50.19 N 12.40 E 5 M Germany 2008-10-14 02:17:06 4.6 38.86 N 23.66 E 38 A Greece 2008-10-14 02:06:39 4.8 38.80 N 23.65 E 10 A Greece 2008-10-14 01:04:25 5.1 5.56 N 124.05 E 498 A Mindanao, Philippines 2008-10-13 21:37:26 4.7 43.44 N 46.47 E 10 A Eastern Caucasus 2008-10-13 17:16:12 5.4 38.82 N 70.58 E 10 A Afghanistan-Tajikistan Border Region 2008-10-13 16:05:27 5.4 39.67 N 73.92 E 38 A Tajikistan-Xinjiang Border Region 2008-10-13 15:56:50 4.9 20.67 S 179.09 W 536 A Fiji Islands Region 2008-10-13 12:07:46 5.0 21.07 S 174.15 W 10 A Tonga Islands Earlier Events Helmholtz-Zentrum Potsdam Deutsches GeoForschungsZentrum - GFZ http://geofon.gfz-potsdam.de/db/eqinfo.php
中国和全球较大地震 发震时间 震级 震中位置 上传时间 20080819 最大震级3.0 四川汶川地震序列(8月19日00时-24时) 8/20/2008 00:20:00 AM 20080818 最大震级4.0 四川汶川地震序列(8月18日00时-24时) 8/19/2008 1:47:00 AM 20080817 最大震级3.1 四川汶川地震序列(8月17日08时-24时) 8/18/2008 12:00:39 AM 20080816 最大震级2.5 四川汶川地震序列(8月16日00时-17日07时) 8/17/2008 07:06:38 AM 20080816 6.0 俄罗斯与蒙古交界 8/16/2008 12:40:38 PM 20080815 最大震级4.9 四川汶川地震序列(8月15日00时-24时) 8/16/2008 00:17:17 AM http://www.eqsn.gov.cn/ Earthquakes Home Audience For Public Earthquakes Recent Earthquakes 08/19/2008 M 6.1, Tonga 08/19/2008 M 5.7, Easter Island region 08/17/2008 M 5.2, Fox Islands, Aleutian Islands, Alaska 08/17/2008 M 5.6, southern Mid-Atlantic Ridge 08/15/2008 M 5.7, Russia-Mongolia border region http://www.iris.edu/hq/audience/public/earthquakes
编者按:有网友提醒,8月8日为上弦,日月潮汐相互抵消,强度最小。请注意观察分析。请检验。 东京及神奈川发生4.5级地震 http://www.sina.com.cn 2008年08月08日14:30 中国新闻网 中新网8月8日电 日本共同社报道,北京时间8日11点57分许,日本整个关东地区发生地震,东京都的八王子堀之内、町田和神奈川县的麻生片平、伊势原均测得震度为4(日本标准)。 据日本气象厅称,本次地震震源位于东京都多摩东部,震源深度约为40公里。 据推算此次地震为里氏4.5级。 http://news.sina.com.cn/w/2008-08-08/143014286482s.shtml 四川什邡和绵竹之间发生4.3级余震 http://www.sina.com.cn 2008年08月08日21:47 新华网 新华网快讯:记者从四川省地震局了解到,8日21时12分四川什邡市和绵竹市之间发生4.3级余震。汶川、北川、青川、成都等地有明显震感。 http://news.sina.com.cn/c/2008-08-08/214716087022.shtml Earthquakes Home Audience For Public Earthquakes Recent Earthquakes 08/08/2008 M 6.0, southern Sumatra, Indonesia 08/07/2008 M 5.8, Andreanof Islands, Aleutian Islands, Alaska 08/07/2008 M 5.2, Fox Islands, Aleutian Islands, Alaska 08/06/2008 M 5.4, Gulf of California 08/06/2008 M 5.9, Sumbawa region, Indonesia http://www.iris.edu/hq/audience/public/earthquakes
 标签: 检验
标签: 检验

![[转载]临床生化检验项目组合](http://image.sciencenet.cn/album/201208/22/130800at6cux9cc6wcy69k.jpg.thumb.jpg)
![[转载]利用SPSS检验数据是否符合正态分布](http://image.sciencenet.cn/album/201112/13/095017p7mj06w4lgmmml77.jpg.thumb.jpg)















