基因个性化果蝇筛选抗癌药物(附原文) 诸平 与癌症患者相似,果蝇化身携带多种基因变化,导致量身定制的治疗,使患者的肿瘤缩小了!這是一個令人興奮的消息。但是,令人遺憾的是研究對象只有一個,既沒有對照,也沒有平行實驗可供參考。 据《科学进展》( Science Advances )网站 2019 年 5 月 22 日发表的一篇论文—— Erdem Bangi, Celina Ang, Peter Smibert, Andrew V. Uzilov, Alexander G. Teague, Yevgeniy Antipin, Rong Chen, Chana Hecht, Nelson Gruszczynski, Wesley J. Yon, Denis Malyshev, Denise Laspina, Isaiah Selkridge, Hope Rainey, Aye S. Moe, Chun Yee Lau, Patricia Taik, Eric Wilck, Aarti Bhardwaj, Max Sung, Sara Kim, Kendra Yum, Robert Sebra, Michael Donovan, Krzysztof Misiukiewicz, Eric E. Schadt, Marshall R. Posner, Ross L. Cagan. A personalized platform identifies trametinib plus zoledronate for a patient with KRAS-mutant metastatic colorectal cancer . Science Advances , 22 May 2019: Vol. 5, no. 5, eaav6528. DOI: 10.1126/sciadv.aav6528 。 A personalized platform identifies trametinib plus zoledronate for a patient wit.pdf 报道了一名身患绝症的结肠癌患者在接受基因修饰果蝇筛选的药物后,接受了一份定制的化疗方案。这种药物组合显著缩小了病人的肿瘤,使其在几个月内保持较小,为进一步研究这种基因个性化的果蝇模型铺平了道路。 没有参与这项研究的美国科罗拉多大学( University of Colorado )的癌症和果蝇专家说:“这是一项令人兴奋的研究成果。因为仅仅涉及到一个病人,所以对于这种方法是否有效,是否适用,当然还有待观察。尽管如此,这也是一个令人兴奋的开始。” 西奈山伊坎医学院( Icahn School of Medicine at Mount Sina )的发育和癌症生物学家、也是此项研究的领导者 Ross Cagan 说,尽管巨大的努力是为了开发新的和更好的化疗药物 , 但是,”一个关键性的挑战就是 , 我们的模型系统转化不易进入诊所进行临床实验 , 因为尚不清楚为什么。” 例如,在结直肠癌中,大约 40% 的患者增加了一种名为 RAS 的癌基因的表达。然而, Ross Cagan 说,专门设计用于抑制 RAS 的药物在临床前实验中效果良好,但在病人身上往往“不成功”。 未参与该项目的英国癌症研究中心的结直肠癌研究人员 Owen Sansom 说:“对于这种 RAS 阳性患者,如果他们的癌症抗拒治疗,在治疗后复发,或已经转移,那就无药可救了。我们亲眼看着患者坐以待毙吗?” Ross Cagan 说,一些最初有希望的药物最终失败的原因有很多。“动物模型通常捕捉一到两个突变,”他解释说,而病人的肿瘤可能有数十或数百个突变,这些突变可能会改变对药物的反应。捕获肿瘤的全部遗传复杂性可以通过在患者手术中移除和培养细胞来实现,然而,这样的体外建模并不能概括身体的生理影响,例如氧合、血液供应以及周围健康组织的影响等。 为了完成遗传和生理的复杂性,病人的细胞可以在老鼠体内生长。但是这些异种移植的动物需要几个月的时间来产生,而且饲养费用昂贵。 看“我的大老鼠( My Mighty Mouse )” 既然老鼠实验不仅费用昂贵,而且周期长。因此, Ross Cagan 的团队转向了一种更便宜、速度更快的动物模型——果蝇——通过对后肠细胞的基因修饰,对人类结直肠癌的几个方面进行了建模,包括过度增殖、肿瘤内分层以及向远处转移的类似转移。研究小组提高了模型的遗传复杂性,至少在一定程度上再现了患者肿瘤的突变情况。 从一名 53 岁结直肠癌 4 期患者的原发肿瘤中提取的 DNA 全外显子组测序显示,存在 100 多个与癌症相关的突变。通过检测这些最可能的驱动突变——那些改变蛋白质功能、影响已知癌症基因或参与与癌症相关通路的突变——研究小组将突变名单缩小到 6 个。他们还确定了患者遗传的一些突变,从中挑选出最可能导致这种疾病的 3 种突变。 在果蝇胚胎的后肠上皮细胞中重现了这 9 种基因异常。然后,这些转基因果蝇被用于筛选 121 种美国食品药品管理局( FDA )批准的药物,这些药物要么是针对癌症,要么是与癌症相关的靶点作用,要么不是针对癌症,但据报道具有抗肿瘤效果。这些药物被混合到昆虫的食物中,科学家们追踪了它们的生存状况。 在不使用药物的情况下,只有不到 20% 的胚胎发育成蛹或成年果蝇,而当对 121 种药物分别进行测试时,存活率并没有上升。然而,进一步涉及药物组合的筛选显示,同时给予两种特定药物——曲米替尼 (trametinib, KRAS 抑制剂 )和双膦酸盐药物(bisphosphonate drug, 用于治疗骨质疏松症 ) ——可将苍蝇存活率提高 30%~60% 。 当这种药物组合给病人时,他的肿瘤缩小了 45% 。在接下来的 11 个月里,肿瘤仍然很小,尽管在前 3 个月之后,它们的大小略有增加,并且出现了 2 个新的肿瘤。在这段时间之后,病人转向了另一种化疗药物,研究人员也就未再继续跟踪病人了。 而导致病人病情变化似乎令人鼓舞 , 但是,未参与此项研究的哈佛医学院的发育遗传学家诺伯特·佩里蒙认为,“ 药物治疗究竟在多大程度上真正对患者病情有所帮助,难以确定。因为实际上没有对照可以参考,只有一个病人 , 我们不知道患者他会对单一药物的治疗效果如何。”不过诺伯特·佩里蒙补充说:“我认为论文的优势在于整体方法,” 诺伯特·佩里蒙他指的是使用基因复杂的体内模型进行药物筛选的好处。 Owen Sansom 补充道:“这是一个非常棒的概念验证研究,展望未来,将会非常有趣……这是否会成为发现新的药物组合的令人兴奋的新方法,还有待深入研究。”更多信息请注意浏览原文或者相关报道。 Abstract Colorectal cancer remains a leading source of cancer mortality worldwide. Initial response is often followed by emergent resistance that is poorly responsive to targeted therapies, reflecting currently undruggable cancer drivers such as KRAS and overall genomic complexity. Here, we report a novel approach to developing a personalized therapy for a patient with treatment-resistant metastatic KRAS-mutant colorectal cancer. An extensive genomic analysis of the tumor’s genomic landscape identified nine key drivers. A transgenic model that altered orthologs of these nine genes in the Drosophila hindgut was developed; a robotics-based screen using this platform identified trametinib plus zoledronate as a candidate treatment combination. Treating the patient led to a significant response: Target and nontarget lesions displayed a strong partial response and remained stable for 11 months. By addressing a disease’s genomic complexity, this personalized approach may provide an alternative treatment option for recalcitrant disease such as KRAS-mutant colorectal cancer.
植物病毒 纳米颗粒传输 药物候选者会使肿瘤缩小 诸平 据美国凯斯西储大学( Case Western Reserve University )2016年6月8日报道,凯斯西储大学和美国麻省理工学院(MIT)的研究人员合作,成功地开发出一种由植物病毒纳米微粒携带的抗肿瘤药物,经过体内实验显示出良好的治疗效果。小鼠模型的三阴乳腺癌使用 phenanthriplatin的效果比顺铂效果更佳。 这种药物作为抗肿瘤药物的候选者,其实 phenanthriplatin就是 在顺铂的基础上进行改进的产物。顺铂和 phenanthriplatin结构式如下: 顺铂结构式 phenanthriplatin结构式 科研人员在实验室发现, phenanthriplatin 直接对抗 肺癌、乳腺癌、骨骼和其他组织癌症癌细胞的能力,要比 传统platins抗癌药物 高40倍!相关研究已经在 ACS Nano 杂志发表。更多信息请浏览: http://phys.org/news/2016-06-drug-candidate-tumor-virus-nanoparticle.html In a pair of firsts, researchers at Case Western Reserve University and Massachusetts Institute of Technology have shown that the drug candidate phenanthriplatin can be more effective than an approved drug in vivo, and that a plant-virus-based carrier successfully delivers a drug in vivo. Triple-negative breast cancer tumors of mice treated with the phenanthriplatin -carrying nanoparticles were four times smaller than those treated either with cisplatin, a common and related chemotherapy drug, or free phenanthriplatin injected intravenously into circulation. The scientists believe the work, reported in the journal ACS Nano , is a promising step toward clinical trials. We may have found the perfect carrier for this particular drug candidate , said Nicole Steinmetz, an assistant professor of biomedical engineering at Case Western Reserve, who has spent 10 years studying the use of plant viruses for medical purposes. She teamed with Stephen J. Lippard, Arthur Amos Noyes Professor of chemistry at MIT, and an expert in biological interactions involving platinum-based chemotherapies. Platinum-based drugs are used to treat more than half of cancer patients receiving chemotherapy. Two of the most commonly used drugs are cisplatin and carboplatin. They form bifunctional cross-links with DNA in cancer cells , which block the DNA from transcribing genes and result in cell death , Lippard explained. Despite widespread use, cisplatin has been shown to cure only testicular cancer, and many cancers have or develop immunity to the drug. Lippard's lab altered cisplatin by replacing a chloride ion with phenanthridine and found that the new molecule also binds to DNA. Instead of forming cross-links, however, phenanthriplatin binds to a single site but still blocks transcription. In fact, his lab found that phenanthriplatin is up to 40 times more potent than traditional platins when tested directly against cancer cells of lung, breast, bone and other tissues. The molecule also appears to avoid defense mechanisms that convey resistance. But when injected into mouse models of cancer, the drug candidate performed no better than standard platins. Lippard realized phenanthriplatin wasn't reaching its target. He had a drug delivery problem. He found a potential solution while visiting Case Western Reserve's campus and heard Steinmetz explain her work investigating tobacco mosaic virus (TMV) for drug delivery more than a year ago. I envisioned that TMV would be the perfect vehicle, Lippard said. So we had a beer and formed a collaboration. The long, thin tobacco mosaic virus nanoparticles are naturals for delivering the drug candidate into tumors, said Steinmetz, who was appointed by the Case Western Reserve School of Medicine. The virus particles, which won't infect humans, are hollow. A central tube about 4 nanometers in diameter runs the length of the shell and the lining carries a negative charge. Phenanthriplatin is about 1 nanometer across and, when treated with silver nitrate, has a strong positive charge. It readily enters and binds to the central lining. The elongated shape of the nanoparticle causes it to tumble along the margins of blood vessels, remain unnoticed by immune cells and pass through the leaky vasculature of tumors and accumulate inside. Little healthy tissue is exposed to the toxic drug. Inside tumors, the nanoparticles gather inside the lysosomal compartments of cancer cells, where they are, in essence, digested. The pH is much lower than in the circulating blood, Steinmetz explained. The shell deteriorates and releases phenanthriplatin. The shell is broken down into proteins and cleared through metabolic or natural cellular processes within a day while the drug candidate starts blocking transcription, leading to greater amounts of cell death through apoptosis than cross-linking platins. The researchers say delivery of the phenanthriplatin into the tumor led to its improved performance over cisplatin or free phenanthriplatin. Lippard and Steinmetz continue to collaborate, investigating use of this system to deliver other drugs or drug candidates, use in other types of cancers, the addition of agents on the exterior of the shell to increase accumulation inside tumors and more.
合成的海绵化合物有望成为抗癌药物 诸平 据 MedicalXpress 网站 2014 年 2 月 7 日 报道,皇家墨尔本理工大学( RMIT University )研究人员指导的博士生丹·巴兰( Dr Dan Balan ),在实验室已经合成出类似于来自天然海生产物中含有的化合物,有望成为癌症治疗药物。 皇家墨尔本理工大学应用科学学院的丹· 巴兰博士( Dr Dan Balan )说:“ 15-aza-Salicylihalamide A 类似物已经证明对多种白血病细胞系是有活性的。 Salicylihalamide A 是一种备受关注的天然海洋产物 , 已经从海生 Haliclona 属海绵中分离得到 , 这种海绵是从距离澳大利亚西南部海岸 18 公里 的罗托尼斯特( Rottnest )岛周围水域采集到的。” Salicylihalamide A 是一种细胞毒素,能够破坏细胞 , 而海生海绵中含有这种物质为其本身提供一种防御作用。巴兰博士说他的目标就是在实验室合成这种化学物质,其结构形式是氮杂 salicylihalamide A 类似物分子 (aza-salicylihalamide A analogue molecule) 。 然后将合成的氮杂 salicylihalamideA 类似物与 NCI-60 白血病细胞系作用 , 观察其抗癌活性。试验结果发现合成的类似物在非常稀的浓度条件下对该细胞系也表现出抗增殖效应。 15-aza-Salicylihalamide A 类似物已被证明对多种癌症类型表现出很强的抗癌活性 , 尤其是对急性早幼粒细胞性白血病 HL-60 的抗癌作用最强。其他研究显示它是一种 空泡 ATP 酶和质子泵 的抑制剂 , 而空泡 ATP 酶和质子泵经常在转移性癌细胞中发现,这是肿瘤细胞通过血流进行迁移。巴兰博士说:“对于药物生产而言,该分子合成简单 , 易于批量生产。 ” 但是他也指出,对于不同癌症的有效性需要进一步的深入研究 , 以便开发适用性更加广泛的抗癌药物。 更多信息请浏览: Synthesised sponge chemical shows promise for cancer ; Sponge Draws Up New Cancer Possibilities ; Synthesis of 15-aza-salicylihalamide A analogues
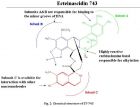
来源于被囊动物红树海鞘 Ecteinascidiaturbinata 的ecteinascidi-743(Et-743,trabectedin)对直肠癌、乳腺癌、肺癌、黑色素瘤等有显著的疗效,2007年10月欧盟已批准该药(商品名Yondelis)用于晚期软组织肿瘤的治疗,成为第一个现代海洋药物。 【药物名称】曲贝替定(Trabectedin), Ecteinascidin 743, NSC-684766, ET-743, Yondelis 【化学名】(1'R,6R,6aR,7R,13S,14S,16R)-5-Acetoxy-6',8,14-trihydroxy-7',9-dimethoxy-4,10,23-trimethyl-1',2',3',4',6a,7,12,13,14,16-decahydro-6H-spiro isoquino benzazocine-20,1'-isoquinolin]-19-one 【CAS登记号】114899-77-3 【结构式】 【分子式】C 39 H 43 N 3 O 11 S 【分子量】761.8447 关于曲贝替定的研究进展以及作用机理请浏览: TRABECTEDIN: A NOVEL MOLECULAR THERAPEUTIC IN CANCER (2013-3-3 15:43:54); A Review of Trabectedin (ET-743): A Unique Mechanism of Action (2013-3-3 15:27:37)
抗血管生成药物被广泛用于癌症治疗。最近,两项分别发表在《Cancer Cell》和《PNAS》杂志的研究揭示,抗血管生成药物会降低其它抗癌药物的效能,增强肿瘤的侵袭性。这两项研究强调,在癌症治疗中要谨慎权衡抗血管生成治疗的利与弊。 《自然》杂志今天发表文章,对这两项研究工作进行了综合评价。 Nature | News Views Cancer: Limitations of therapies exposed Oriol Casanovas Nature Volume: 484 , Pages: 44–46 Date published: (05 April 2012) Published online 04 April 2012 Certain drugs that are used to treat cancer affect blood-vessel formation in tumours. But it seems that these antiangiogenic drugs can reduce the efficiency of other anticancer agents and increase the tumours' aggressiveness. Tumour growth depends on angiogenesis, the formation of new blood vessels, to ensure a continuous supply of oxygen and nutrients. That is why antiangiogenic agents are used to treat certain cancers, either alone or in combination with traditional cytotoxic drugs. However, the mechanistic details of how these combination therapies work are far from clear, and accumulating evidence is exposing their limitations. Writing in Cancer Cell , Van der Veldt et al . 1 report that angiogenesis inhibitors can decrease the delivery of cytotoxic drugs to tumours in patients, and hence hinder the drugs' therapeutic benefits. And in a paper published in Proceedings of the National Academy of Sciences , Conley et al . 2 find that tumours can adapt to antiangiogenic therapy by accumulating particularly aggressive cells. The main target of current antiangiogenic agents is a protein called vascular endothelial growth factor (VEGF), which has a central role in angiogenesis. Although it has been known for several years that VEGF inhibitors provide additional antitumour effects when combined with cytotoxic drugs ( Fig. 1a ), the underlying mechanism has been a mystery since the early positive results of drug-combination trials 3 . The most widespread explanation for such a mechanism, the 'vascular normalization' theory, was proposed in 2001. According to this theory 4 , 5 , antiangiogenic therapy induces structural and functional changes in tumour blood vessels — which have abnormal features — to make them more similar to normal vessels and, as a result, blood flow is increased and cytotoxic drugs can more easily enter the tumour. Figure 1: Drawbacks of anticancer therapies. a , Certain cancers are treated with antiangiogenic drugs (which affect blood-vessel formation), either alone or in combination with cytotoxic agents that inhibit the growth of cancer cells. Some of these cells (green) are particularly dangerous because they can be more resistant to cytotoxic therapy than the other tumour cells and can spread to other organs to seed new tumours. b,c , A decreased blood supply to the tumour, which is the main benefit from antiangiogenic therapy, is also the basis for the therapy's limitations. b , It can reduce the distribution of cytotoxic agents in the tumour, and hence their efficacy. Van der Veldt et al . 1 report one such undesirable outcome in patients with non-small-cell lung cancer. c , By reducing oxygen levels in the tumour, antiangiogenic drugs can induce the accumulation of more aggressive cells that have an increased capacity to spread to other organs. Conley et al . 2 document this phenomenon in mouse models of breast cancer in the absence of a cytotoxic agent. To test the theory in a clinical setting, Van der Veldt and colleagues 1 studied the uptake and retention of a cytotoxic drug (docetaxel) in 10 patients with advanced-stage non-small-cell lung cancer (NSCLC). By using radiolabelled docetaxel together with a sensitive imaging method (positron emission tomography), the authors demonstrate that VEGF inhibition with a drug called bevacizumab induces a fast and sustained decrease — not increase — in the penetration of both water and docetaxel in the tumours ( Fig. 1b ). These results contrast with those of previous studies in patients with rectal cancer 6 and in patients with glioblastoma (a brain tumour) 7 , which showed that bevacizumab treatment induces vascular normalization and increased glucose uptake in the tumours. However, glucose uptake by tumour cells does not, in my opinion, necessarily correlate with cytotoxic-drug delivery and penetration into tumours. The discrepancies between the authors' observations 1 and previous results 6 , 7 could also be due to differences in blood-vessel networks and in the response to angiogenesis inhibitors between the three cancer types, as it is known that these agents can affect blood vessels in different ways in different tissues 7 , 8 . In any case, the finding 1 that, at least in patients with NSCLC, antiangiogenic therapy does not improve cytotoxic drug delivery to tumours — but rather has the opposite effect — exposes a perturbing drawback to such treatments. Indeed, this could be the cause of the modest benefits of these combination therapies in NSCLC and other tumour types 9 . Such a potential shortcoming could be circumvented by optimizing the scheduling of the therapeutic agents. For example, rather than administering both types of drug to a patient during the same period, treatment with blood-distributed cytotoxic agents could be followed by antiangiogenic therapy. Other limitations of the use of angiogenesis inhibitors derive from the fact that tumours are highly adaptable. Although their ability to become resistant to cytotoxic drugs and radiation — another common anticancer therapy — has long been known, it was initially postulated 10 that antiangiogenic drugs would not suffer from the same problem because they target blood vessels rather than tumour cells. Yet preclinical and clinical evidence 11 , 12 has revealed that tumours can indeed adapt and become resistant to antiangiogenic therapy. As if that was not bad enough, angiogenesis inhibitors have been shown 13 , 14 to make some tumours more aggressive in animal models ( Fig. 1c ). To explore this issue, Conley and co-workers 2 implanted human cancer cells (derived from established breast- cancer cell lines) in mice. They then treated the animals with the antiangiogenic agents sunitinib — which inhibits VEGF's main cell-surface receptors — and bevacizumab. The treatment induced an accumulation of certain cancer cells that expressed the enzyme aldehyde dehydrogenase and that, like cancer progenitor cells, could initiate tumours when reimplanted in other mice. Similar cell populations have been described in tissue samples from patients with inflammatory breast cancer 15 and in glioblastoma in mice given combination therapies 16 . Conley et al . 2 go on to delineate a possible cellular and molecular mechanism for the increased aggressiveness and spread capacity of tumours treated with antiangiogenic drugs. They find that the drugs, by inducing oxygen deficiency (hypoxia) in the tumours, activate not only a hypoxia-response program but also the Akt/β-catenin signalling pathway, which regulates cell growth and adhesion between cells. This pathway has been previously implicated in the regulation of breast-cancer progenitor cells 17 . The authors suggest that the drug-induced hypoxia response activates the Akt/β-catenin pathway, which in turn stimulates the growth of specific, more aggressive, cancer-cell populations. How could this drawback of antiangiogenic therapies be overcome? One possibility would be to combine angiogenesis inhibitors with drugs that suppress the cancer cells' response to hypoxia, or with inhibitors of the Akt/β-catenin pathway. Another alternative could be the use of molecules such as modified semaphorin proteins 18 , which can exert dual (or multiple) anticancer effects by simultaneously targeting angiogenesis and blocking tumour spread. Overall, the papers by Van der Veldt et al . 1 and Conley et al . 2 emphasize the need for a carefully balanced evaluation of the benefits and limitations of antiangiogenic therapies. As mentioned above, such treatments could be improved by sequential scheduling of cytotoxic and antiangiogenic drugs, or by smarter combinations of these drugs with others targeting progenitor-cell pathways. In any case, despite many open questions, there is hope that an understanding of the therapies' weaknesses will translate into therapeutic gains. References Van der Veldt, A. A. M. et al . Cancer Cell 21 , 82 – 91 ( 2012 ). CAS PubMed Article Show context Conley, S. J. et al . Proc. Natl Acad. Sci. USA 109 , 2784 – 2789 ( 2012 ). PubMed Article Show context Hurwitz, H. I. et al . J. Clin. Oncol. 23 , 3502 – 3508 ( 2005 ). CAS ISI PubMed Article Show context Jain, R. K. Nature Med. 7 , 987 – 989 ( 2001 ). Article Show context Jain, R. K. Science 307 , 58 – 62 ( 2005 ). CAS ISI PubMed Article Show context Willett, C. G. et al . Nature Med. 10 , 145 – 147 ( 2004 ). Article Show context Batchelor, T. T. et al . Cancer Cell 11 , 83 – 95 ( 2007 ). CAS ISI PubMed Article Show context Kamba, T. et al . Am. J. Physiol. Heart Circ. Physiol. 290 , H560 – H576 ( 2006 ). CAS ISI PubMed Article Show context Sandler, A. et al . N. Engl. J. Med. 355 , 2542 – 2550 ( 2006 ). CAS ISI PubMed Article Show context Boehm, T. , Folkman, J. , Browder, T. O'Reilly, M. S. Nature 390 , 404 – 407 ( 1997 ). CAS ISI PubMed Article Show context Bergers, G. Hanahan, D. Nature Rev. Cancer 8 , 592 – 603 ( 2008 ). Article Show context Rini, B. I. Atkins, M. B. Lancet Oncol. 10 , 992 – 1000 ( 2009 ). CAS ISI PubMed Article Show context Pàez-Ribes, M. et al . Cancer Cell 15 , 220 – 231 ( 2009 ). CAS ISI PubMed Article Show context Ebos, J. M. L. et al . Cancer Cell 15 , 232 – 239 ( 2009 ). CAS ISI PubMed Article Show context Charafe-Jauffret, E. et al . Clin. Cancer Res. 16 , 45 – 55 ( 2010 ). CAS ISI PubMed Article Show context Folkins, C. et al . Cancer Res. 67 , 3560 – 3564 ( 2007 ). CAS ISI PubMed Article Show context Korkaya, H. et al . PLoS Biol. 7 , e1000121 ( 2009 ). CAS PubMed Article Show context Casazza, A. et al . EMBO Mol. Med. 4 , 234 – 250 ( 2012 ). CAS PubMed Article Show context http://www.nature.com/nature/journal/v484/n7392/full/484044a.html#/references
 标签: 抗癌药物
标签: 抗癌药物