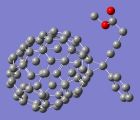
90年后,科学家揭示了苯的结构 诸平 Fig. 1 DVMS structures for benzene. a Voronoi site for the RHF/6-31G(d) wavefunction. The electron positions of an arbitrary spin are shown as small yellow spheres. b Cross sections through the wavefunction around the Voronoi site in a C–C bonding electrons are shown as blue lobes. C–H bonds are shown in grey. c. Voronoi site showing staggered spins. The electron positions of each spin are respectively shown as small yellow and green spheres. d. Cross sections around the Voronoi site in c. The two spins of the C–C bonding electrons are shown in blue and red. C–H bonds are shown in grey. Credit:Nature Communications(2020). DOI: 10.1038/s41467-020-15039-9 苯的结构是一名不知名的奥地利中学教师约瑟夫·劳施密特( Josef Loschmidt in full Johann Joseph Loschmidt , born May 15, 1821, Putschin, Bohemia, Austrian Empire —died July 8, 1895, Vienna , Austria ),早在 1861 年就已经得知苯环的结构了,这是在著名化学家凯库勒( Friedrich August Kekulé , 1829-1896 )梦里幻想苯环结构之前的事。因为 六元芳环在生物学中无处不在, 它是 DNA 和蛋白质以及木质生物质和石油 的组成部分 。芳族环和芳族结构在星际空间中观察到,在一般情况下,被认为是遍及星际介质。共轭六元环也是石墨烯的基本组成部分,石墨烯是具有惊人电子特性的材料。因此,对其结构的研究意义重大。 据澳大利亚研究委员会 ( Australian Research Council , ARC ) 激子科学卓越中心( ARC Centre of Excellence in Exciton Science )2020年 3 月 5 日提供的消息, 90 年后,科学家揭示了苯的结构。相关研究结果于 2020 年 3 月 5 日已经在《自然通讯》( Nature Communications )杂志网站发表—— Yu Liu , Phil Kilby , Terry J. Frankcombe , Timothy W. Schmidt . The electronic structure of benzene from a tiling of the correlated 126-dimensional wavefunction, Nature Communications , 2020, volume11, Articlenumber:1210. DOI: 10.1038/s41467-020-15039-9 图1是苯的DVMS结构。a. 是RHF/6-31G(d)波函数的Voronoi站点(Voronoi site)。任意自旋的电子位置显示为黄色小球。b. 在C-C键电子中,Voronoi站点周围波函数的横截面显示为蓝色瓣;C-H键显示为灰色。c. Voronoi站点显示交错旋转。每个自旋的电子位置分别显示为黄色和绿色小球。d. c中Voronoi站点周围波函数的横截面。C–C键合电子的两个自旋以蓝色和红色显示。C-H键显示为灰色。图片来源: Nature Communications (2020) . DOI: 10.1038/s41467-020-15039-9 .顺便介绍一下 Voronoi 图, Voronoi 图是一种重要的几何结构,它既是一种行之有效的空间剖分和聚类方法,又具有骨架的特性。它按照站点( sites )集合中元素的最近邻属性将空间划分成许多单元区域。在不同应用背景下,根据生成空间、测量距离以及站点等定义条件的不同,又产生了不同类型的 Voronoi 图。 ARC激子科学卓越中心、澳大利亚新南威尔士大学(University of New South Wales,UNSW)和澳大利亚联邦科学与工业研究组织(Commonwealth Scientific and Industrial Research Organization, CSIRO)之间的合作解决了化学的基本谜团之一,其结果可能会对太阳能电池,有机发光二极管和其他下一代技术的未来设计产生影响。 自20世纪30年代以来,化学界内部就苯的基本电子结构展开了激烈的争论。近年来,这种争论变得更加紧迫,因为包含六个碳原子和六个氢原子的苯(C 6 H 6 )是许多光电材料的基本组成部分,这正在彻底改变可再生能源和电信技术。扁平的六角环也是DNA、蛋白质、木材和石油的组成部分。围绕苯分子结构的争论之所以出现,是因为尽管它的原子成分很少,但电子以不仅包括四个维度(four dimensions,如我们日常的“大”世界),而且包括126个维度的状态存在。 到目前为止,分析复杂的系统被证明是不可能的,这意味着无法发现苯电子的精确行为。这就是一个问题,因为如果没有这些信息,该分子在技术应用中的稳定性将永远无法被完全理解。 但是,现在,由ARC卓越科学中心和新南威尔士大学悉尼分校(UNSW Sydney)的蒂莫西·施密特( Timothy Schmidt )领导的科学家,成功地揭开了谜底-结果令人惊讶。他们发表在《自然通讯》杂志上论文,解开了126个维度的谜团。 蒂莫西·施密特教授与来自UNSW和CSIRO的Data61( CSIRO’s Data61 )的同事们一起,对苯分子应用了一种基于动态Voronoi大都会采样(dynamic Voronoi Metropolis sampling, DVMS)的复杂算法,以便在所有126个维度上绘制其波函数。 解决复杂问题的关键是由合作者CSIRO的Data61菲尔·基尔比博士(Dr. Phil Kilby)开发的新数学算法。该算法允许科学家将维空间划分为等效的“平铺”(equivalent tiles),每个平铺对应于电子位置的排列。 科学家特别感兴趣的是理解电子的“自旋”。所有电子都有自旋 , 这是产生磁力以及其他基本力的特性,但它们如何相互作用是从发光二极管到量子计算的广泛技术之基础。 蒂莫西·施密特教授说:“我们发现的结果非常令人惊讶。”“既有具有上自旋双键的电子,也有具有下自旋单键的电子,反之亦然。”“这并非化学家对苯的想法。本质上,它通过使彼此排斥的电子相互排斥,从而降低了分子的能量,使其更稳定。” 来自Data61的合作者菲尔·基尔比博士补充说:“尽管是针对这种化学背景而进行的开发,但是我们为“与约束条件匹配”而开发的算法 , 其应用范围并非仅仅而已,它也可以应用于从工作人员名册到肾脏交换计划的广泛领域。”更多信息请注意浏览原文或者相关报道。如理论家们最终证明,“弯箭”能说明化学反应的真相( Theoreticians finally prove that 'curly arrows' tell the truth about chemical reactions )—— Yu Liu , Philip Kilby , Terry J. Frankcombe , Timothy W. Schmidt .Calculating curly arrows from ab initio wavefunctions. Nature Communications , 2018,Volume9, Articlenumber:1436. DOI: 10.1038/s41467-018-03860-2 Abstract The electronic structure of benzene is a battleground for competing viewpoints of electronic structure, with valence bond theory localising electrons within superimposed resonance structures, and molecular orbital theory describing delocalised electrons. But, the interpretation of electronic structure in terms of orbitals ignores that the wavefunction is anti-symmetric upon interchange of like-spins. Furthermore, molecular orbitals do not provide an intuitive description of electron correlation. Here we show that the 126-dimensional electronic wavefunction of benzene can be partitioned into tiles related by permutation of like-spins. Employing correlated wavefunctions, these tiles are projected onto the three dimensions of each electron to reveal the superposition of Kekulé structures. But, opposing spins favour the occupancy of alternate Kekulé structures. This result succinctly describes the principal effect of electron correlation in benzene and underlines that electrons will not be spatially paired when it is energetically advantageous to avoid one another.
 标签: 计算化学
标签: 计算化学