博文
Discovery Studio官方教程(Help-Tutorials) 构建基于分子共同特征的药效团模型
|
目的: 通过此教程,了解Discovery Studio中构建基于分子共同特征的药效团的操作方法及结果分析。
所需功能和模块:Discovery Studio Client,DS Catalyst Conformation,DS Catalyst Hypothesis,DS Catalyst Score。
所需数据文件:5HT2c_ligands.sd,sulfonamide5HT2c_ligands.sd,sampledrugs20.sd。
所需时间:30分钟
介绍
Common Feature Pharmacophore Generation protocol (HipHop) 用于发现一系列配体小分子所共有的化学特征,并基于这些共同特性结构的比对叠合自动生成药效团模型,用户可以使用共有的特征药效团去搜索化合物数据库来寻找可能的先导分子。基于分子共同特征的药效团模型同样也可以用于探索一系列具有相似活性但结构却不同或者结构柔性较大的分子的构效关系。
此教程以6个活性配体小分子所构成的训练集来构建基于分子共同特征的药效团模型,继而用于先导化合物的发现。本教程包括以下步骤:
· 基于分子共同特征的药效团模型的构建(训练集)
· 基于分子共同特征的药效团模型的验证(测试集)
· 先导化合物的发现(数据库的筛选)
Common Feature Pharmacophore的构建
1. 训练集分子的准备
本教程采用一系列已知的5-HT2c配体(1,2)来构建一个基于分子共同特征的药效团模型用于先导化合物的发现。5-HT2c受体属于GPCR超家族。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开5HT2c_ligands.sd。
在表格浏览器中可以看到一共有6行,代表了6个分子。这些分子的Principal和MaxOmitFeat属性都已事先定义。若无定义,则选择表格浏览器中剩下列的heading,鼠标右键点击选择Add Attributes,打开Add Attributes对话框,添加这两个属性。

本教程假设所有分子都是活性分子并且视其中某几个分子为参考分子。如果所选取的分子有着截然不同的药效特征类型和尺寸,那么Principal和MaxOmitFeat属性设置的不同对最终构建的药效团模型影响较大。
2. 药效团特征元素的选取
肉眼观察这几个活性化合物,选出可能的药效团特征元素,或者根据Feature Mappinp 工具来完成该步骤(如下)。
点击表格浏览器中的按钮,在图形窗口中显示化合物5HT2c_ligand1。再在图形窗口右击选择Show All,显示所有活性化合物的结构。
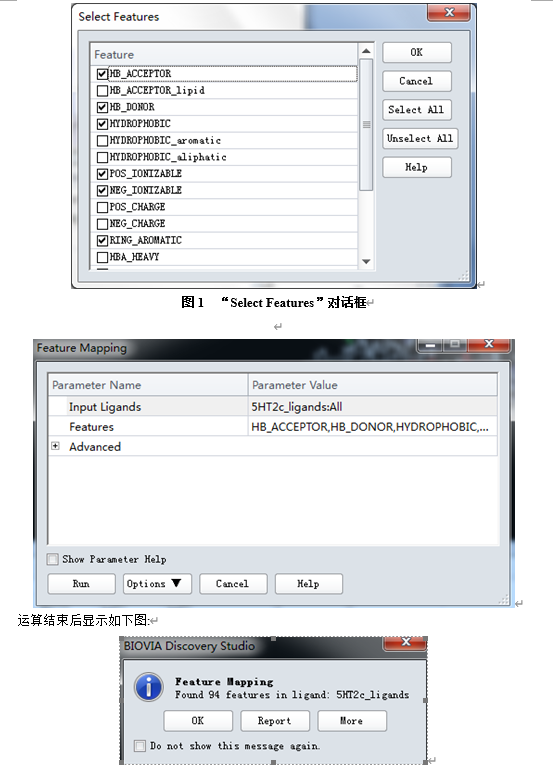
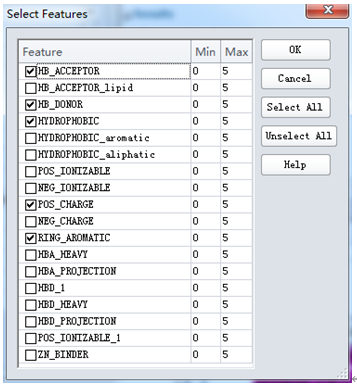
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Edit and Cluster Features,在工具面板中单击Feature Mapping打开Feature Mapping对话框,点击Features右边按钮,打开Select Features对话框(图1),选择所要匹配的特征元素,此处设为默认值。
点击Run运行该功能,并等待计算完成。
该操作可以识别出所有已显示化合物中所有所选特征元素可能的位置。
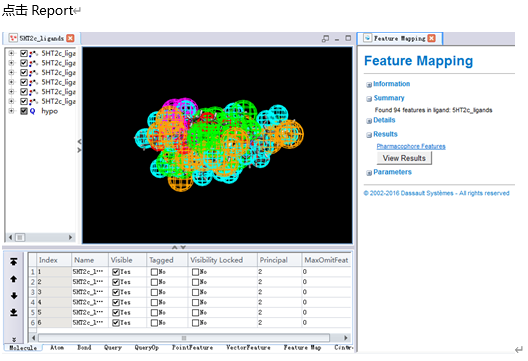
鼠标左键点击药效团分子窗口,激活该窗口,在Edit and Cluster Features工具面板中单击Current Features查看训练集分子所表征的药效团特征元素的所有类型。
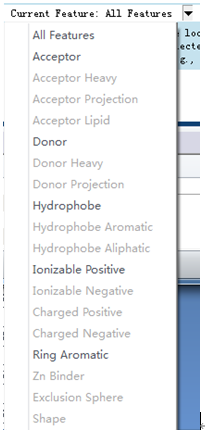
结果显示这六个化合物都包含有有疏水中心(Hydrophobe)、氢键供体(Donor)、氢键受体(Acceptor)、正电离子中心(Ionizable Positive)及芳香环中心(Ring Aromatic)这五种特征元素的类型。
3. Common Feature Pharmacophore的构建
在工具浏览器(Tools Explorer)中,展开Pharmacophores | Create Pharmacophores Automatically,单击Common Feature Pharmacophore Generation。
流程对应参数在参数浏览器中打开。
设置Input Ligands为5HT2c_ligands:All。
点击Feature右边 按钮,打开Select Features对话框。
按钮,打开Select Features对话框。
勾选POS_IONIZABLE和RING_AROMATIC左边的复选框,点击OK。(下图)
在默认设置基础上添加上一个正电离子特征POS_IONIZABLE,一个芳香环特征RING_AROMATIC。
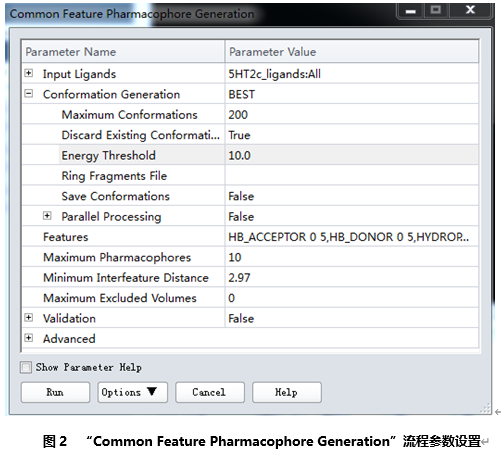
展开Conformation Generation参数组,点击Conformation Generation右边的栅格,下拉列表中选择BEST,构象上限数Maximum Conformation设为200,能量阈值Energy Threshold设置为10,其他参数默认。(图2)
对每个小分子产生最多200个构象用以表征小分子的构象空间。其中只有同最低能构象的能量差值在能量阈值10kcal/mol之内的构象被保留。
点击Run运行作业。
在弹出的窗口点击Background。
等待作业完成。该作业大概需要5分钟。
从菜单中选择Window | Close All,关闭所有窗口。
药效团结果分析
1. 查看结果
作业完成后,展开作业浏览器(Jobs Explorer)中该任务并点击Report链接,在Html窗口中打开Report页面。
从Summary一栏可知,此次运算一共产生了10个药效团。在Details一栏中我们可以了解到每个药效团更为详细的信息。Results一栏罗列了各个结果的链接。Parameters一栏显示了此次操作所采用的各参数。
这10个药效团模型是根据训练集分子与药效团模型的匹配度以及模型本身的稀有度来排名的。排名第一的模型未必是最好的模型,因此需要对这10个模型进行综合分析。
2. 查看药效团与训练集分子的匹配情况
在Report页面中展开Results,点击View Aligned Ligands 01链接查看训练集分子同排名第一的药效团的匹配情况。
点击表格浏览器中的 键,查看排名第一的分子同药效团的匹配,继而点击
键,查看排名第一的分子同药效团的匹配,继而点击 键和
键和 键,可以查看到每个分子与该药效团的匹配情况。
键,可以查看到每个分子与该药效团的匹配情况。
在表格浏览器中,注意查看5HT2c_ligand4和5HT2c_ligand5的FitValue值。
回到Report页面,点击View Aligned Ligands 10链接查看训练集分子与排名第十的药效团的匹配情况。
在此次叠合中,5HT2c_ligand5的FitValue比5HT2c_ligand4高,且匹配度要好。
分析Common Feature Pharmacophore的结果时需要考察所有药效团模型同训练集分子的匹配情况,了解每个模型是如何解释训练集分子的。
回到Report页面,展开Details一栏。(图3)
该表格罗列了十个药效团模型的结果参数。查看该表格可以快速了解到十个药效团模型之间的不同。
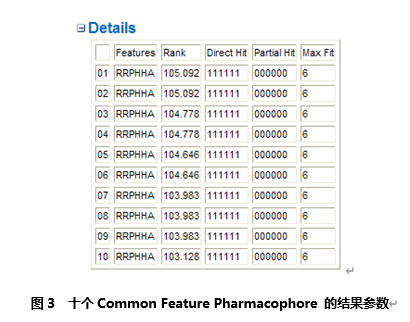
表格中每一行代表一个药效团。
每一栏的含义如下:(以模型01为例)
Features:RRPHHA此药效团含有2个芳环中心,1个正电离子中心,2个疏水特征,1个氢键受体特征;
Rank: 105.092此药效团的打分值为105.092;
DH: 111111此药效团药效特征与6个小分子均可匹配;
PH: 000000此药效团与6个小分子部分匹配的药效特征数目为0;
Max Fit: 6最大匹配值为6,即6个药效特征均可匹配。
3. 查看药效团与测试集分子的匹配情况
最终决定选取哪一个药效团模型用于表征训练集分子以及用于寻找潜在的新型5-HT2c配体分子,是一个主观的过程。因此,可以基于对训练集化合物同关键化学特征间叠合情况的分析。同时也可以采用测试集化合物来验证确定。
注:同样可以通过设置上述protocol中的Validation参数来自动完成验证过程。产生的每一个药效团模型都会同诱饵分子数据库(含actives和inactives)进行匹配并最终自动生成ROC曲线显示于Report页面。
本教程中所采用的测试集分子包含了负性分子,对模型作了一个简单的验证,从而确定最终的药效团模型用于识别潜在的新型小分子骨架。
从菜单中选择Window | Close All,关闭所有窗口。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开sulfonamide5HT2c_ligands.sd。在文件浏览器(Files Explorer)中,通过File|Insert From|File...将sampledrugs20.sd打开至同一窗口中。
第一个文件包含了18个已知5-HT2c活性的磺胺类分子[Garzya et al.,2007]。第二个文件包含了20个同6个训练集分子非常类似(基于Lipinski和FCFP_6属性)的配体小分子,视这20个化合物为非活性化合物(理想情况应该是这些分子确实是负性对照分子,即确实不能与受体结合)。
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Search, Screen and Profile,点击Ligand Profiler。
流程对应参数在参数浏览器中打开。
该流程可以将多个分子同多个药效团模型快速匹配。
设置Input Ligands为sulfonamide5HT2c_ligands:All。
点击Input File Pharmacophores参数,选择Browse,找到之前运行Common Feature Pharmacophore构建流程所得到的Output文件。SHIFT-选取所有模型。
展开Conformation Generation参数组,点击Conformation Generation右边的栅格,下拉列表中选择BEST,构象上限数Maximum Conformation设为200,能量阈值Energy Threshold设为10,点击Save Conformations右边的栅格,下拉列表中选择True。
展开Advanced参数组,点击Scale Fit Values右边的栅格,下拉列表中选择False。点击Save Aligned Ligands右边的栅格,下拉列表中选择True。
点击Options选取Save Protocol Settings保存参数设置。
若下次运行该流程时想要采用原始参数的设置则可以点击Options选取Restore Original Settings。
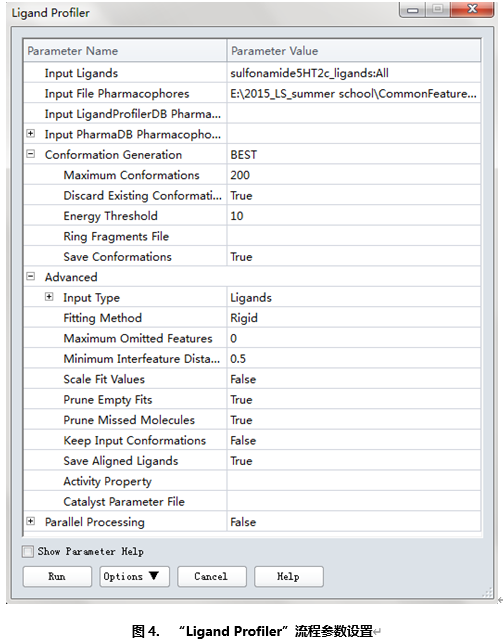
其余参数使用默认值。(图4)
点击Run运行作业。
点击Background等待作业完成。
该作业大概需要9min。
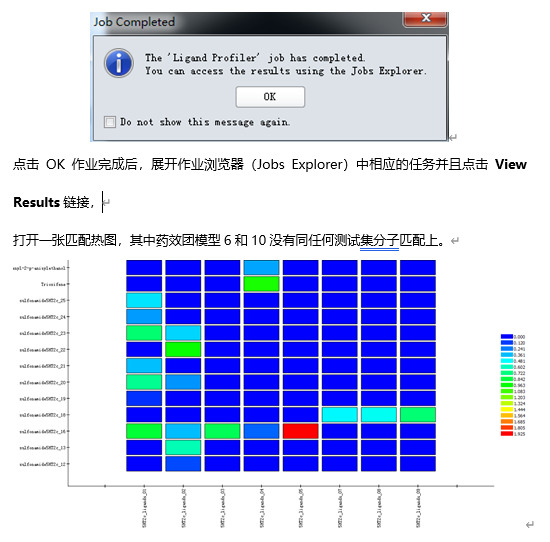
展开作业浏览器(Jobs Explorer)中相应的任务并且点击5HT2c_ligands_05_view.pl,在图形窗口中右击选取Show All查看所有同模型01匹配上的测试集分子同模型01的匹配情况。
结果显示只有1个配体分子同药效团模型05相匹配,且FitValue值都偏低。造成这一现象的原因可能是Maximum Omitted Features参数设成了0,即不允许任何一个药效团特征元素不被匹配上。该参数设为-1表示考虑所有药效团特征元素的子集。
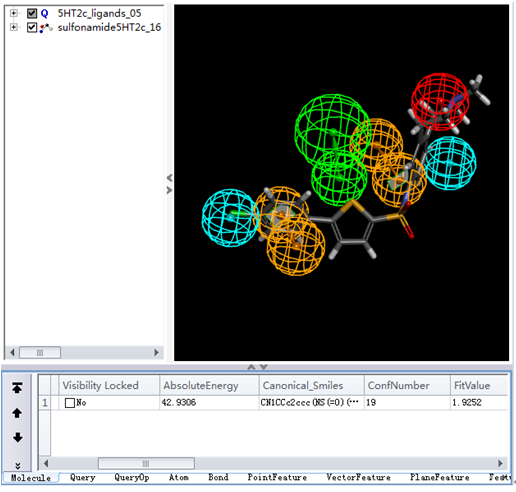
再次点击Ligand Profiler。
点击Input Ligands右侧栅格,下拉列表中选取Browse,找到上一步运行得到的Output文件,选取sulfonamide5HT2c_ligands_BEST.sd。
将Conformation Generation改为NONE。其余参数与之前一样.
Conformation Generation设为NONE,是因为上一步已经保存了所产生的构象(即Save Conformations设为TRUE),不需要重复。
展开Advanced参数组,将Maximum Omitted Features右边的数值改为-1以允许部分匹配。
其余参数使用默认值。(图5)
点击Run运行作业。
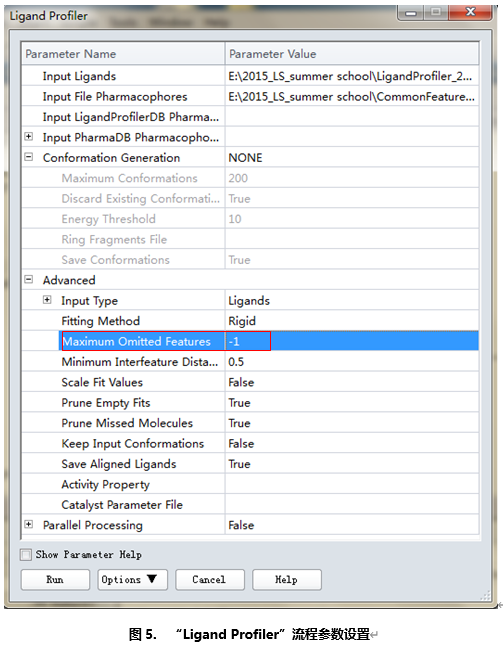
点击Background等待作业完成。
该作业大概需要5min。
作业完成后,双击作业浏览器(Jobs Explorer)中相应的行,打开Report页面。
展开Results,点击View Results按钮。
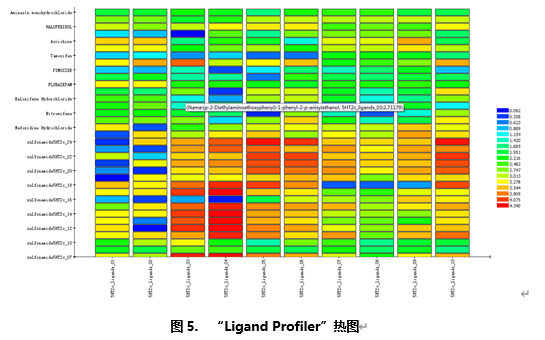
自动生成一张表格和一个热图(图6)。
在表格浏览器中,有18个活性磺胺类分子和17个非活性配体分子。
热图中横坐标第一列有几个区域显示为蓝色,表示同第一个药效团模型匹配的某些配体小分子fit值比较低。同样地,在药效团模型5HT2c_ligands_04一列有几个区域显示为橙色和红色,表示同5HT2c_ligands_04药效团模型匹配的某些配体小分子fit值比较高。
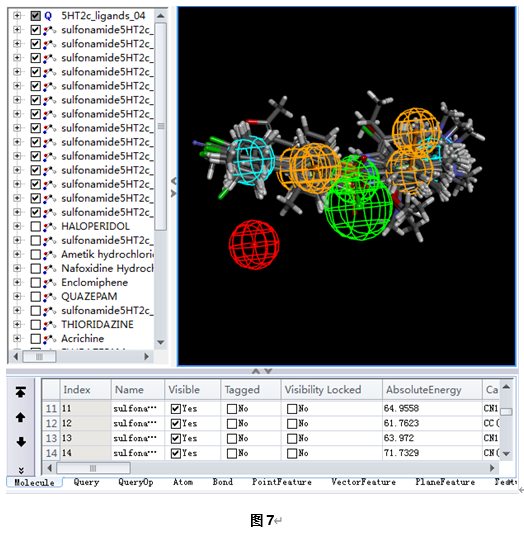
展开Results,点击5HT2c_ligands_04_view.pl。
在表格浏览器中,化合物按FitValue从高到低排列,前14个小分子都是活性化合物。
在Hierachy窗口中,SHIFT-选取前14个分子,在Graphics窗口中右击选取Show。
结果显示这14个分子的叠合情况有所改善,但所有的分子都没有同正电离子特征匹配。
先导化合物的发现(数据库筛选)
采用上述得到的3D药效团模型对MiniMaybridge化合物数据库进行虚拟筛选,以期发现潜在的新型先导化合物。
1. 采用药效团进行数据库筛选
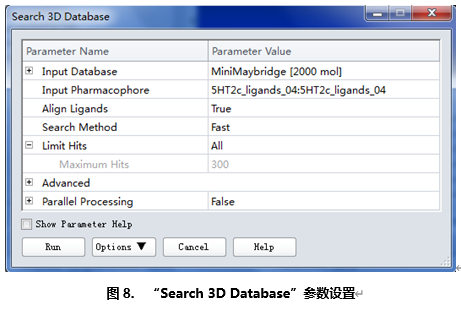
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Search, Screen and Profile,点击Search 3D Database。
流程对应参数在参数浏览器中打开。
点击Input Database后面栅格,下拉菜单中选择MiniMaybridge[2000 mol]。
此操作表示在MiniMaybridge数据库中进行搜索,该数据库中含有2000个小分子。
在Input Pharmacophore后选择5HT2c_ligands_04: 5HT2c_ligands_04。
此操作表示搜索可以与5HT2c_ligands_04药效团模型相匹配的小分子。
其余参数设诶默认设置。(图8)
点击Run运行作业。点击Backgroud等待作业完成。从菜单中选择Window | Close All,关闭所有窗口。
2. 查看筛选结果
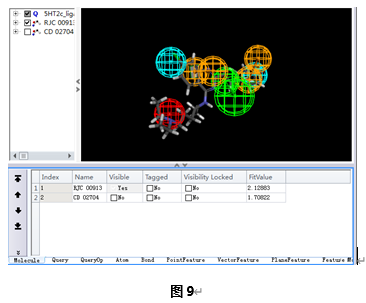
双击作业浏览器(Jobs Explorer)窗口中相应的行,打开Report页面点击ViewResults按钮可以查看小分子结构是否同原训练集分子及活性测试集分子有结构上的差别。
表格浏览器(Data Table)中的FitValue栏目中的数值可以评价小分子与药效团匹配的情况,FitValue值越高,则小分子与药效团匹配的越好。可以发现筛选到的这两个小分子从结构上来说,无论是同训练集分子还是测试集中活性分子相比,都有较大差异。

https://m.sciencenet.cn/blog-3536821-1362025.html
上一篇:Discovery Studio官方教程(Help-Tutorials) 构建具有活性预测能力的3D QSAR药效团
下一篇:Discovery Studio官方教程(Help-Tutorials) 拉伸动力学计算结合自由能