博文
磷酸化激酶规模化分析新技术
 精选
精选
|
蛋白质磷酸化就是将磷酸基团选择性地添加到蛋白质中,是一种对生命至关重要的调节机制。磷酸化修饰可以让蛋白质局部亲水性发生改变,从而对蛋白质的空间结构和相互作用发生影响。
磷酸化失调与阿尔茨海默病、癌症和糖尿病等疾病有关。细胞内催化磷酸化的酶,称为激酶,是药物的主要靶标,因此了解它们的调节作用可以提供新的治疗机会。我们使用质谱法鉴定和定量磷酸化的能力的进步导致人类蛋白质(统称为磷酸化蛋白质组)中已知磷酸化位点的数量迅速增加,从世纪之交的数百个增加到今天的10万多个。然而,将这些位点与其相关的激酶联系起来是一个费力的过程。约翰逊等发表在《自然》杂志的研究朝着解决这个问题迈出重要一步,描述一个全面的资源,定义了一个几乎所有人类激酶成员的潜在底物。
磷酸化蛋白质组非常复杂,由数百种蛋白激酶及其数万个底物紧密互连的网络组成。它们共同形成细胞信号网络,可以像微处理器一样发挥作用,通过编码、处理和整合细胞信息,并以无数细胞过程的形式调节输出,从基因表达到细胞分裂。这种能力之所以成为可能,只是因为不同的激酶对许多可能的蛋白质底物具有不同的特异性。
激酶的特异性源于许多外在和内在因素。外在因素包括激酶及其底物是在同一细胞类型中表达还是在细胞的同一部分中表达,以及与其他分子(如支架蛋白)的相互作用。内在因素来自激酶和底物的生化和结构特性。例如,磷酸化位点附近存在带电或大块氨基酸残基可能会促进或阻碍激酶磷酸化给定蛋白质的能力。内在特异性导致激酶具有首选基序 - 围绕磷酸化位点的理想残基序列。
Johnson等使用称为位置扫描肽阵列分析的无细胞技术确定几乎所有靶向氨基酸丝氨酸和苏氨酸的激酶的内在底物特异性,这种激酶约占人类细胞磷酸化位点的99%。该方法涉及筛选303种人激酶的文库,以确定每种激酶将磷酸基团转移到数百种不同短链氨基酸的中央丝氨酸或苏氨酸残基的能力(图1)。值得注意的是,作者发现几乎三分之二的磷酸化位点可以分配给少数激酶之一。
Targets mapped for almost all human kinase enzymes (jd314.vip) 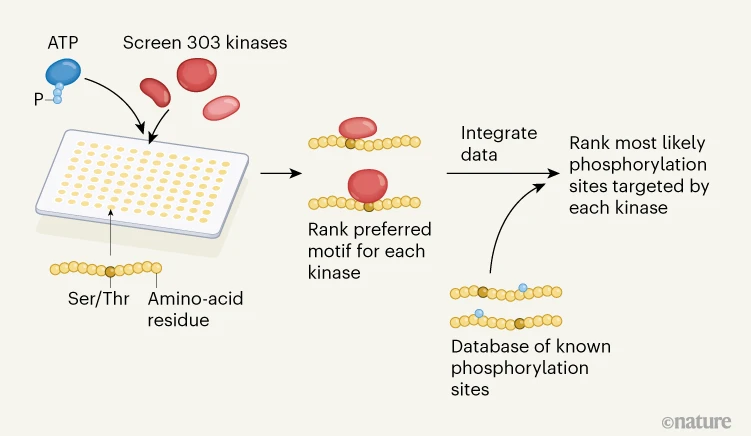
图1 |激酶-酶底物特异性的大规模分析。
该研究还强调了“负选择性”在定义底物选择方面的重要性。正如一些激酶在其靶标的特定位置偏爱某些氨基酸残基一样,反之亦然 - 针对此类残基的选择 - 也发生了。这已经为一些激酶所知,但作者表明,这是一种一般性质,驱动了“kinome”(人类激酶的完整库)的整体底物选择性的大部分。
通过系统地定义大多数人类丝氨酸/苏氨酸激酶的内在底物特异性,作者对这些酶进行了广泛的比较。激酶超家族以前被分为100多个家族,主要是基于其催化结构域的序列比较。假设内在底物特异性遵循这些家族分组,因为催化域的序列(以及扩展的结构)决定了底物的可及性。但约翰逊及其同事的发现导致作者将这些激酶重新分类为至少38个基于基序的类别。他们发现来自不同系统发育家族的激酶汇聚在相似的底物序列特异性上,并且他们定义了激酶中决定性的关键特异性氨基酸残基,这些残基负责一些(但不是全部)观察到的选择性。
由于预测蛋白质结构计算工具AlphaFold等的发展,结构模型的可用性不断增加,将允许将激酶及其推定底物的结构信息纳入特异性预测中。对于这些预测的准确性至关重要的是将酶本身的修饰包含在激酶结构(以及所有蛋白质结构)模型中,因为许多这些酶仅在其催化区域本身磷酸化时才具有活性。
这些新定义的激酶特异性也可用于预测100多种“暗”激酶的功能 - 那些没有任何报道底物的激酶。组装每种激酶的潜在底物并将某些特征(例如它们表达的组织,它们的功能以及如何调节)整合到各种生物学环境中可以揭示这些研究不足的酶的生物学作用。这种可能性应该激励研究人员冒险“进入黑暗”,以更好地表征这些难以捉摸的蛋白质。这将是特别有益的,因为生物医学研究倾向于关注某些“受青睐”的蛋白质,即使这些蛋白质不一定比其他任何蛋白质更重要,无论是对于生物还是医学研究。学者因为熟悉,生物信息学因为全面而研究这些反复被研究的蛋白。这导致了许多分子生物学研究的大量而毫无价值。
今天,质谱法可以测量微小生物样品中蛋白质的数以万计的变化12.然而,尽管由此产生了大量信息,但研究人员经常根据仅5%的磷酸化位点的反应来解释细胞信号网络的变化,这些位点的激酶已被鉴定。为了解决这一缺点,许多研究已着手使用有限的可用实验数据预测某些(但不是全部)激酶的底物。相比之下,Johnson等人使用他们的实验衍生数据对最有可能作用于大多数人类磷酸化蛋白质组的激酶进行计算排序(图1)。研究人员现在可以在分析细胞信号数据集时使用这些预测,以获得生物样本中发生的潜在网络活动的更广泛视图。
随着生物医学领域继续以加速的速度和规模生成生物分子的轮廓,将这些数据情境化的方法的进步至关重要。约翰逊及其同事的数据库将使研究人员能够在各种生物学环境中预测感兴趣分子的调节介质。该方法可以扩展到酪氨酸激酶(此处未包括的最大激酶家族)的分析,或其他类型的修饰,例如添加泛素或乙酰基,甚至扩展到修饰之间的相互作用,提供更全面的细胞信号传导视图。
虽然仅内在方面可以很好地解析激酶的潜在底物,但激酶特异性也受到其他因素的影响。理想情况下,这些因素 - 包括蛋白质 - 蛋白质相互作用,组织和细胞特异性表达以及细胞定位 - 可以纳入细胞信号网络的拓扑模型中。然而,这将需要关于许多细胞类型和生物学背景的大量数据。幸运的是,基于质谱的蛋白质组学正在崛起以应对这一挑战。我们绘制调节激酶的方式的持续改进将增强我们解释细胞信号语言的能力。
https://m.sciencenet.cn/blog-41174-1371601.html
上一篇:试验表明结肠镜检查不会预防死亡风险,怎么回事?
下一篇:人工智能撰写论文的时代来了!