开启微生物暗物质大门
“暗物质”,有奶便是娘,但是“奶”在哪?
所谓微生物“暗物质”,我指的是那些无法被培养的细菌。想必你一定见过涂布、划线得单克隆。但实际上,不能够在培养基里单独培养的菌的种类还有很多,空气中、皮肤上、肠道里、口腔中等等特殊的环境下生存的微生物,不是都能够通过牛肉膏、蛋白胨什么的轻易培养进而分离出来的,而因为共生关系(比如地衣就是真菌和藻类的共生体)使得单独分离纯化出某一种菌,更加困难。
太多未知细菌必需生存环境的缘故,微生物学家并不能成功培养所有的细菌。传统的微生物学研究,研究的都只是浩瀚微生物里的冰山一角。于是如何更好地鉴定、研究这些无法通过培养得到的细菌,这算是微生物学领域的一段佳话。
16s rRNA,微生物鉴定利器
基因组学(genomics)成于微生物研究。通过培养得到细菌,分离细菌DNA,分析16s rRNA。16s rRNA作为保守序列,广泛用来细菌分类。当年的第三界古生菌(Archaea)的问世,就是拜赐与rRNA。不同种类的细菌在16s rRNA上会有不同差异,是天然的物种标签。
16s rRNA被用来鉴定细菌,显著卓越,鉴定了广泛的微生物。[1]
但是很显然,人们真正感兴趣的是细菌的基因!但是要研究基因,必须需要大量的DNA样品。对于无法培养的细菌种类,就无法扩增得到单纯的DNA样品。这样的研究思路,无疑是条死路。
Metagenomics:大杂烩
Metagenomics一词最早1998年就被提出来。研究的样品直接从环境中得到(比如肠道、口腔)。既然不能得到单独种类菌落,那么就索性一不做二不休,不再按照传统思路、不再琢磨如何培养特殊菌的单菌落,而是研究某个特殊环境下,测序得到的genome的大杂烩。分析这个大杂烩的成分组成以及成分组成随环境变化而发生的变化。
Metagenomics相比于Genomics有两个飞跃:
第一,不再琢磨如何培养特殊菌的单菌落。也就是不再纠结这个物种的菌落喝什么奶。
第二,既然微生物存在种间关系,常常发生的1+1>2的关系。与其一开始就锁紧眉头一味研究单种菌,还不如就干脆研究更有意义的种间关系、种內关系。(当然随后第二步就是解偶关系并分清主次关系)
比如人类肠道微生物就登陆《nature》2010年3月4日封面,被誉为人类的第二基因组。[3]从丹麦和西班牙124位健康的、超重的和肥胖的成年人以及炎症患者提取的人肠道微生物菌落的一个基因目录。比如大家喝的益力多,就是基于这么个科研背景(本人并不看好益力多,仔细留意一下产品成分里给的是什么普通的菌就知道了;而且以国内的运输条件来看,养在小瓶子的菌早死了;即使没死,被胃酸泡过后就都废了)
MDA:满足求知欲
Metagenomics研究的是基因组大杂烩,但是这道杂烩里主菜是什么?如果想要弄清楚,metagenomics是断然办不到的。终究是要回归到单种菌落的研究中去!于是老问题就又再一次出现:这genome怎么得到?
科学研究是个螺旋上升的过程,为了解决这个问题,MDA诞生了![2]下图就是MDA扩增原理。
MDA(multiple displacement amplification)的意义在于,为司法鉴定提供帮助!让单细胞测序提供可能!只需要极少的DNA模板就可以扩增产物。都知道一般扩增DNA使用的方法是PCR手段,但是PCR手段需要的DNA初始浓度至少是纳克(ng)级别的,所以对于超小量的样品,PCR无解。而MDA就有用武之地了。
对于某一种菌,得到了它的单细胞,虽然不能放在平板上克隆繁殖,但是通过MDA就可以实现其基因的测序,一窥其基因奥秘。
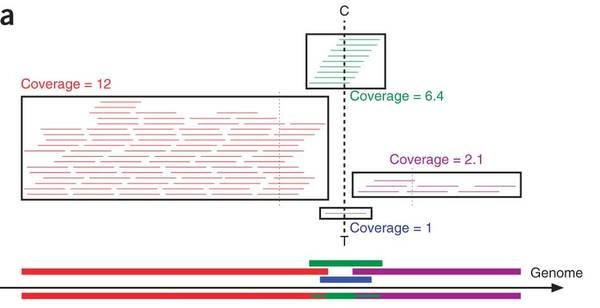
讲到这里,科普也就差不多了。测序后的reads组装,MDA手段得到的reads让一般的组装算法会崩溃掉的。因为MDA可以说是一种简单的复制器,因此reads覆盖度在整个genome上看会有bias。高表达的基因会狂多reads覆盖,从而显示的是深度覆盖;而低表达的基因会少的可怜reads,从而显示低深度覆盖。一般的算法或者流程(比如著名的Bowtie等等)把reads组装成contig时,会对reads覆盖度有个筛选。低于某个深度(比如4X)的区域就会被忽略。如图,紫色和蓝色的就会被筛去,而留下红色与绿色。
所以,问题就出现了,那些明明属于基因组的基因区域,只是因为MDA的技术误差原因,就被忽略了!组装出的genome不完整!
所以2011年又是来自Craig J Venter研究中心的人(和UCSD),[5] 整出了一个算法,就是专门做单细胞测序并de novo组装!使用的程序名称是Velvet-SC(Velvet modified for Single Cell reads)和EULER-SR (Error Correction components)。[4] 就完成了对单细胞genome的测序,并且de novo组装。
我之所以一再提de novo组装genome,是因为相对于通过pair end(PE)关系来做的reads组装,de novo组装会更靠谱一些。通过PE关系做的genome组装,genome size比真实结果一定会小!而de novo组装的结果,相对而言,因为reads彼此之间是类似于瓦片一样的重叠关系,所以得到的genome更能够真实还原原来的genome。
这一篇文章,两个大亮点:
第一:single-cell的测序
第二:de novo组装
end && reference:
[1] Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious
[2] http://en.wikipedia.org/wiki/Multiple_displacement_amplification
[3] A human gut microbial gene catalogue established by metagenomic sequencing
[4] http://bix.ucsd.edu/projects/singlecell/
[5] Efficient de novo assembly of single-cell bacterial genomes from short-read data sets