博文
【CELL】compass利用单细胞转录组+流平衡分析预测细胞代谢状态,促进免疫细胞代谢研究
|
细胞代谢调控正常细胞功能以及多种疾病状态的病理生理。最近,免疫细胞代谢研究(immunometabolism)成为一个研究热点,揭示了包括抗病毒免疫、自身免疫和抗肿瘤反应在内的炎症代谢调节。然而,由于代谢网络的规模和复杂性,某个代谢的扰动(perturbation)能够产生级联效应(cascade effects)并改变代谢网络中看似较远的部分或者让经典通路“弯道超车”。因此,需要在系统水平上观测特定反应/酶,以理解代谢及其在疾病中的异常调控。在CELL文章《Metabolic modeling of single Th17 cells reveals regulators of autoimmunity》中作者提出了一种基于单细胞RNA测序和流平衡分析(flux balance analysis)描述细胞代谢状态的算法 – compass(https://github.com/YosefLab/Compass)。
流平衡分析(flux balance analysis)将代谢网络拓扑学和化学计量学知识翻译成数学对象,并用在代谢流预测上。提供了一种非常有用的场景:根据单细胞基因表达谱或者传统RNA-seq基因表达谱研究细胞的代谢异质性。
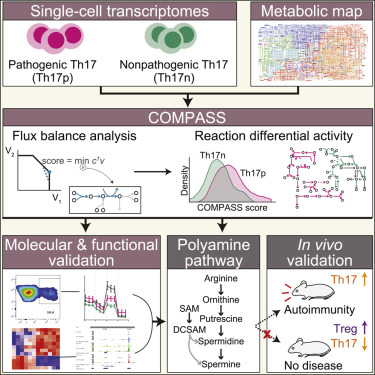
图1. COMPASS算法
如图1所示,使用单细胞转录组(Th17p细胞和Th17n细胞)和代谢模型作为COMPASS的输入,经过流平衡分析,找到了两群细胞间异常的代谢通路(图2)及相关的反应/酶,并进行了分子和功能验证,以及小鼠体内验证。
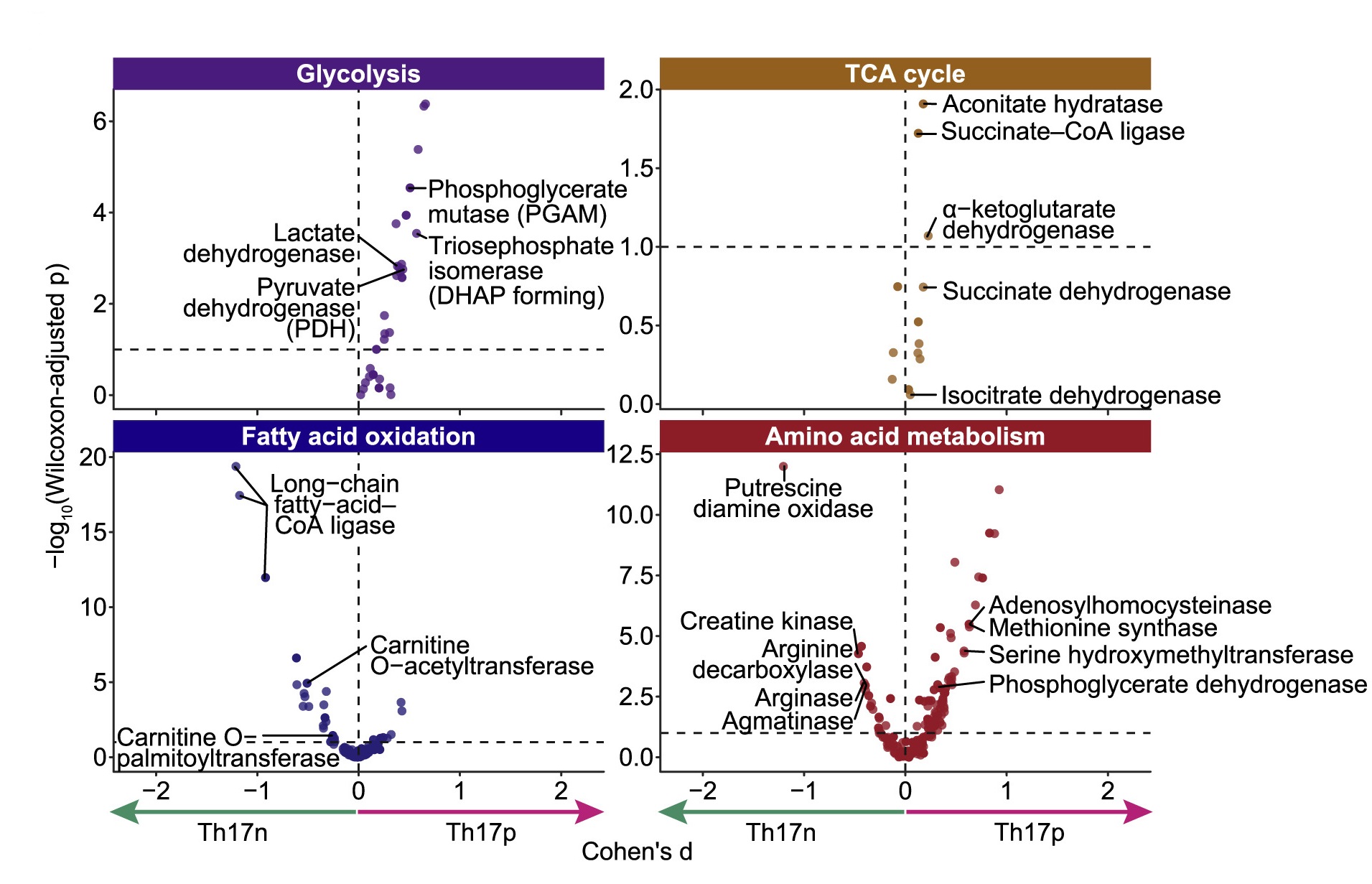 图2.4个差异通路
图2.4个差异通路
如图2所示:左上:糖酵解通路(Glycolysis)在Th17p细胞中更加活跃(反应/酶几乎都分布在虚线右侧);右上:三羧酸循环(TCA cycle)中大部分反应/酶在Th17p细胞中更活跃。左下:脂肪酸氧化的3个酶在Th17n细胞中更活跃;右下:氨基酸代谢中,丝氨酸羟甲基化转移酶在Th17p细胞中更活跃,而精氨酸脱羧酶在Th17n细胞中更活跃。
看到这里,大家应该对文章有了基本的理解。细胞代谢网络太复杂,难以研究,然而我们手上不是有现成的一堆单细胞/bulk RNA-seq的表达数据么,我们可以通过大量样品和严格的假设检验以酶的mRNA表达为桥梁来研究细胞代谢,将基因表达-代谢反应/酶活性-细胞代谢状态联系起来。例如我们知道了某反应在癌细胞中活跃,就可以通过抑制剂阻断(或者平衡)这个反应,改变免疫细胞的代谢状态,从而达到治疗癌症的目的。
1.安装
Compass是用python编写的,需要python 3环境,numpy,scipy,matplotlib等包。
由于计算量太大,还需要一个IBM CPLEX Optimization Studio优化器(学术用户可以注册下载,注意版本)
conda 个python3环境
pip install numpy, scipy, matplotlib
下载IBM CPLEX Optimization Studio(需要学术版)
./xxx.bin 装到software目录(自己指定),一路enter或者yes
cd /home/software/CPLEX_Studio129/cplex/python/3.6/x86-64_linux
python setup.py install
python -m pip install git+https://github.com/yoseflab/Compass.git --upgrade
compass -h 测试安装是否成功
2.运行
输入:表达矩阵(tpm,cpm等,一个基因一行,一个样品一列),单细胞的话配上genes和sample文件。运行速度很慢,可以多线程加速
 图3. Expression.csv输入
图3. Expression.csv输入
compass --data expression.csv --num-processes 10 --species homo_sapiens (常规转录组)
compass --data-mtx expression.mtx genes.tsv sample_names.tsv --num-processes 10 --lambda 0.25 --species homo_sapiens (单细胞)
3.结果
运行完上面的命令后,会在当前目录生成reaction.csv文件(图4)。其中的值是反应罚分(reaction penalties),高得分表示该反应的可能性较低。
 图4. reaction.csv输出
图4. reaction.csv输出
4. 后处理
步骤3获得的是每个反应在每个样品中的反应罚分,我们需要根据Recon2 meta信息(图5)将A列的反应编码转成人们可读的代谢通路名字,将反应罚分进行转化(转成数值大的表示反应活性高,加1,取-log),然后使用wilcoxon或者Cohen’d进行差异分析。
 图5. 后处理需要的3个文件
图5. 后处理需要的3个文件
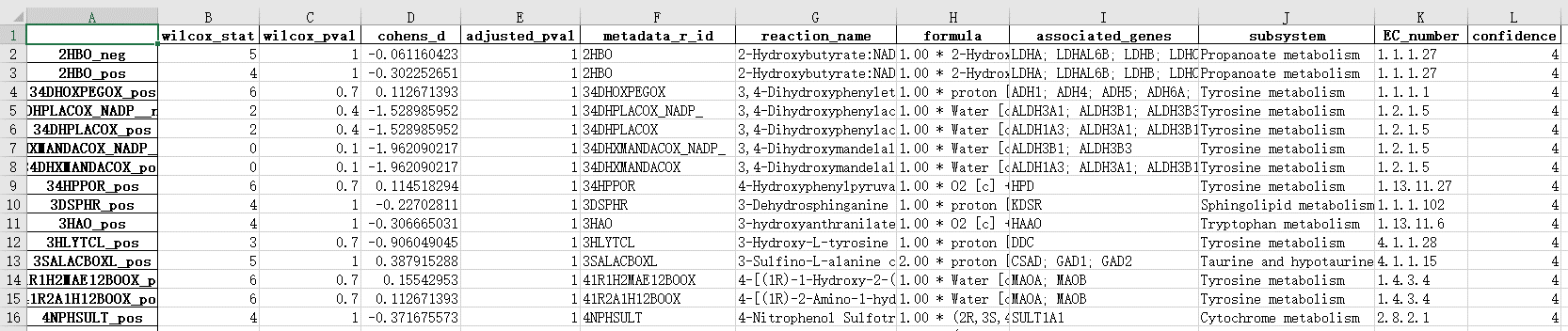 图6. 最终结果表格,根据这个表格寻找更兴趣的反应/代谢,绘图
图6. 最终结果表格,根据这个表格寻找更兴趣的反应/代谢,绘图
各列说明:
A:reaction编码, “_pos” 表示化学反应往右侧,“_neg”表示反应向左侧
B/C/E:wilcoxon统计结果
D:cohens_d: cohen’s d用于计算两组均值差异的效应量, d = 0.2(效果小) ; d = 0.5(效果中) ; d = 0.8(效果大)。供参考
F:reaction编码
G:reaction名字
H:reaction公式
I:相关基因
J:reaction所处的subsystem(例如酪氨酸代谢)
E:酶的EC号
L:置信度, 4 = most confident; 0 = unassigned confidence
注:1,由于单细胞计算量非常大,作者提供了一个micropooling脚本,将细胞划分为簇,以簇的平均值代表这类细胞,然后进行分析。
2,You may also apply Compass to bulk transcriptome data sets (e.g. bulk RNA-seq or microarray data sets) if there are enough observations (samples) to gain statistical power。可以用在常规转录组,甚至芯片数据集上。
有单细胞测序数据、常规转录组测序数据、芯片数据、代谢组数据的小伙伴可以试试这款软件。又打开了一个新的免疫代谢相关发文思路!
微生信助力高分文章,用户63000+,引用830+
https://m.sciencenet.cn/blog-707141-1367902.html
上一篇:在线绘制富集分析多组气泡图和单细胞分析marker基因矩阵气泡图
下一篇:免费在线绘制高颜值,带填充的连贯堆叠柱状图