博文
Discovery Studio官方教程(Help-Tutorials) 构建具有活性预测能力的3D QSAR药效团
|
目的: 通过此教程,了解Discovery Studio中构建具有活性预测能力的药效团的操作方法及结果分析。
所需功能和模块:Discovery Studio Client,DS Catalyst Conformation,DS Catalyst Hypothesis,DS Catalyst Score。
所需数据文件:serm_ligands.sd,test_serm_ligands.sd。
所需时间:30分钟
介绍
3D Quantitative Structure-Activity Relationship (QSAR) Pharmacophore Generation protocol可以基于一系列针对特定生物靶标具有明确活性数值的化合物构建出具有活性预测能力的药效团模型。该算法首先构建得到活性分子能共享而非活性分子则不能共享的初始药效团模型,然后再经过模拟退火进行模型的进一步优化。最终构建得到的模型可以预测化合物的活性,以及指导化合物的优化以提高其活性。
此教程中我们采用一系列已知活性值的训练集分子来构建一个3D QSAR药效团模型。
本教程包括以下步骤:
l 3D QSAR药效团的构建(训练集)
l 药效团结果分析
u 叠合&成本分析(cost analysis)
u 测试集
l 化合物的设计改造
3D QSAR药效团的构建
1. 训练集分子的准备
本教程采用一系列选择性的雌激素受体调节剂(SERMs)[Broqi et al., 2009]来构建一个3D QSAR药效团模型用于预测新型小分子的活性。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开serm_ligands.sd。
在表格浏览器中可以看到一共有24行,代表了24个分子。这些分子的名字(Name)、活性(Activ,可以为IC50或Ki值,且活性跨度需跨越4个数量级)和活性不确定度(Uncert)都已事先输入。Activ值为SERMs对MCF7人体胸腺恶性肿瘤细胞增生的抑制活性IC50(μM)值,Uncert值为1.5(该值的设定取自于文献[Broqi et al., 2009])。
在表格浏览器中,点击 键和
键和 键,在3D Window中观察各个分子的结构特征。
键,在3D Window中观察各个分子的结构特征。
2. 药效团特征元素的选取
在表格浏览器中,右击鼠标并选择Color By Activity。
双击Activ一栏,使24个化合物按活性从高到低排列(即活性值从低到高)。
根据药效团构建算法排名最靠前的2个化合物被定义为活性化合物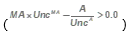 ,其所代表的两行显示为浅绿色。排名最靠后的7个化合物则被定义为非活性化合物
,其所代表的两行显示为浅绿色。排名最靠后的7个化合物则被定义为非活性化合物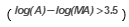 ,其所代表的行显示为浅粉色。观察前两个活性化合物,选出可能的药效团特征元素,或者根据Feature Mapping来完成该步骤(如下)。
,其所代表的行显示为浅粉色。观察前两个活性化合物,选出可能的药效团特征元素,或者根据Feature Mapping来完成该步骤(如下)。
点击表格浏览器中的 按钮,显示活性最高的化合物(serm-40)。
按钮,显示活性最高的化合物(serm-40)。
在表格浏览器中勾选活性排名第二高化合物的visible复选框(serm-34)。
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Edit and Cluster Features,单击Feature Mapping,打开参数面板。
设置Input Ligands参数为serm_ligands:Visible。
在参数面板中点击Features右边 …按钮,打开Select Features对话框,选择所要匹配的特征元素,此处设为默认值。
…按钮,打开Select Features对话框,选择所要匹配的特征元素,此处设为默认值。
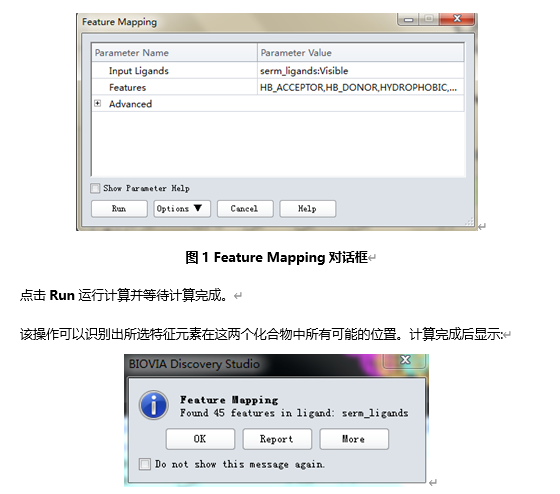
点击OK,在Edit and Cluster Features工具面板中单击Current Features:All Features查看训练集分子所表征的药效团特征元素的所有类型。
结果显示这两个化合物都包含有有疏水中心(Hydrophobe)、氢键供体(Donor)、氢键受体(Acceptor)、正电离子中心(Ionizable Positive)及芳香环中心(Ring Aromatic)这五种特征元素的类型。在构建药效团模型时采用Hydrophobic Aromatic特征取代Ring Aromatic特征,因为前者限制条件少,只定义了一个位点,没有后者的平面和矢量限制。
一旦药效团模型构建好以后,通常需要依据模型对于测试集分子活性预测能力来评判药效团模型的优劣。在3D QSAR Pharmacophore Generation这一protocol中可以自动运行这一验证步骤。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开test_serm_ligands.sd。
3. 3D QSAR药效团的构建
在工具浏览器(Tools Explorer)中,展开Pharmacophores | Create Pharmacophores Automatically,单击3D QSAR Pharmacophore Generation。
流程对应参数在参数浏览器中打开。
设置Input Ligands为serm_ligands:All。
展开Validation参数组,并设置为True。
设置Input Test Ligands为test_serm_ligands:All。
设置Minimum Interfeature Distance为1.5。
展开Conformation Generation参数组,点击Conformation Generation右边的栅格,下拉列表中选择FAST,能量阈值Energy Threshold设置为10,其他参数默认。(图3)
对每个小分子产生最多255个构象用以表征小分子的构象空间。其中只有能量值在能量阈值10kcal/mol之内的构象被保留。
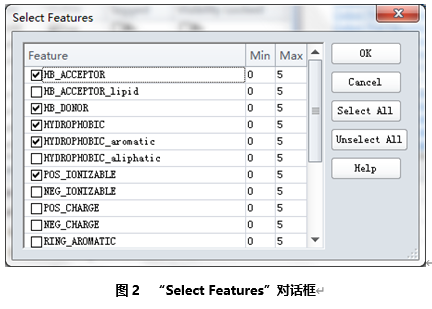
点击Feature右边![]() 按钮,打开Select Features对话框。
按钮,打开Select Features对话框。
勾选POS_IONIZABLE和HYDROPHOBIC_aromatic左边的复选框,点击OK。(图2)
在默认设置基础上添加上一个正电离子特征POS_IONIZABLE,一个芳香环特征HYDROPHOBIC_aromatic。
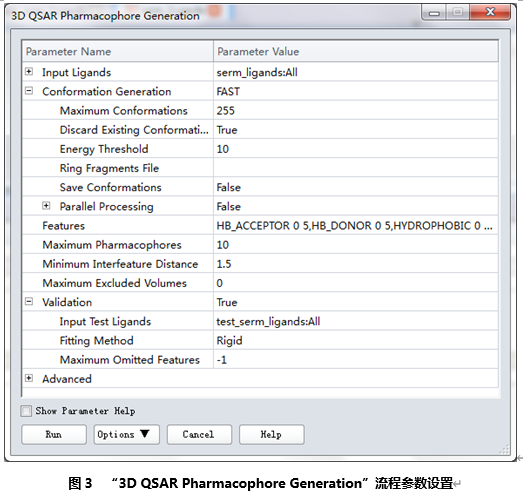
点击Run运行作业。
点击Background等待作业完成。
该作业大概需要6min。
药效团结果分析
选择一个最好的3D QSAR药效团模型不仅仅只是考虑药效团的排名(打分排名根据cost值的高低)。排名第一的药效团模型并不一定就是最好的模型。因此,除了分析模型的cost值,还需要考察训练集分子与模型的匹配情况从而考察模型是如何解释不同的小分子具有不同的活性值。此外最重要的,是用已知活性值的测试集分子来验证模型对训练集以外的分子活性值的预测能力。
1. 查看Report页面
作业完成后,双击作业浏览器(Jobs Explorer)中相应的行,打开Report页面。
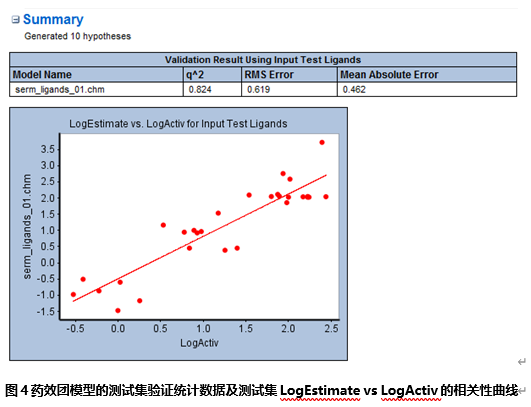
从Summary一栏可知,此次运算一共产生了10个药效团,且包含测试集验证统计数据及测试集LogEstimate vs LogActiv的相关性曲线(图4)。在Details一栏中我们可以了解到每个药效团更为详细的信息。Results一栏罗列了各结果的链接。Parameters一栏显示了此次操作所采用的各参数。
利用上述测试集验证信息可以先快速的选取出预测活性能力较好的模型,此处为模型5、8最好。接着即可关注这些模型的cost值。
2. 查看药效团与训练集分子的匹配情况
查看训练集分子与所有模型的匹配情况相对比较耗时。该教程中用户可以简单查看训练集分子和药效团模型如何进行匹配。主要考察依据是相较非活性分子,活性分子同模型中匹配的特征元素应更多。也就是说,非活性分子应该缺失某一重要的特征元素,或特征元素存在但在空间上不能进行正确取向。因此,选取药效团模型的一个重要指标即基于训练集分子和药效团模型的匹配情况能够解释对应训练集分子的生物活性。
在Report页面中展开Results,点击View Aligned Ligands 01链接查看训练集分子同排名第一的药效团的匹配情况。
在表格浏览器中,可以看到有一列名为Estimate的栏目,此列为使用该药效团对每个化合物的活性预测值。此列在制作Common Feature的药效团中是看不到的。
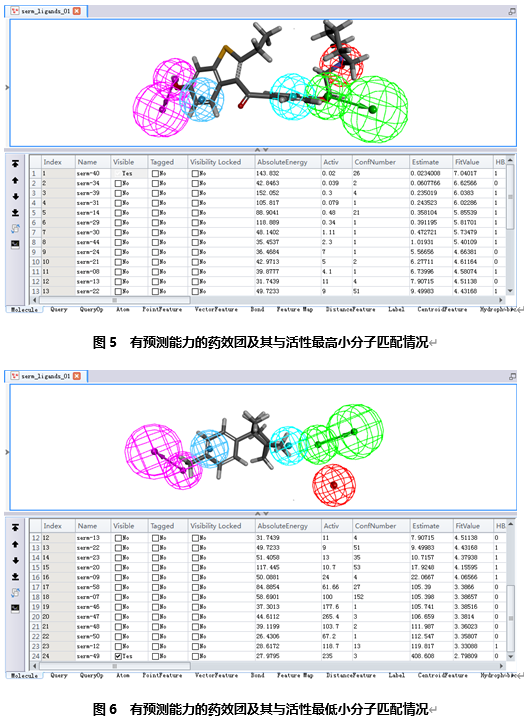
点击 键显示预测活性最高的化合物(serm-40)同该药效团匹配的情况。(图5)
键显示预测活性最高的化合物(serm-40)同该药效团匹配的情况。(图5)
该化合物的实验活性值为0.02μM,预测活性值则为0.023μM。该化合物同药效团的所有特征元素都匹配的很好。
点击 键显示预测活性最低的化合物(serm-49)同该药效团匹配的情况。(图6)
键显示预测活性最低的化合物(serm-49)同该药效团匹配的情况。(图6)
该化合物的实验活性值为235μM,预测活性值则为408μM,仍在同一数量级。该化合物只能同药效团中五个特征元素中的两个特征元素匹配上。
点击 键和
键和 键,可以查看到每个分子与该药效团的匹配情况。
键,可以查看到每个分子与该药效团的匹配情况。
接下去查看药效团模型的log file。
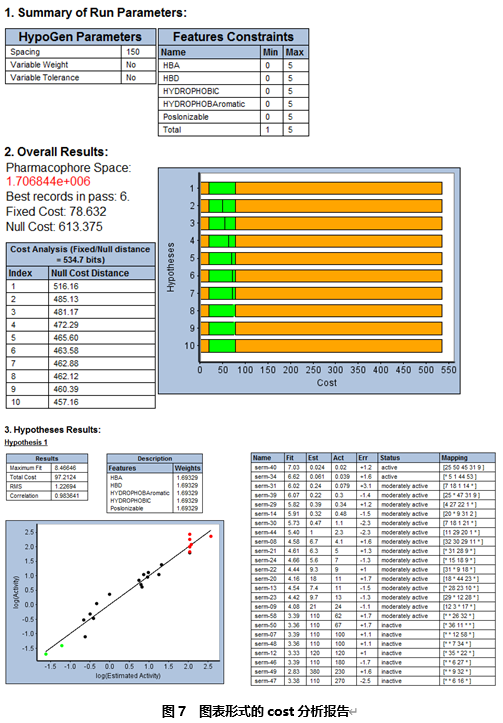
展开作业浏览器(Jobs Explorer)中相应的行,点击Detailed Analysis Report打开图表形式的cost分析报告。(图7)
该报告主要由三部分组成:
l 重要的操作参数及选取的特征元素;
l 十个药效团模型的cost分析摘要;
l 每个药效团模型同训练集分子匹配情况的图表表示。
△cost(Null cost - Total cost)是评价一个药效团模型的重要指标,该值为40-60,表明该模型的置信区间为75%-90%,该值小于40,则模型的置信区间降为50%以下。
对第三部分图表的简单分析表明第一个药效团模型对训练集化合物活性的预测能力最好,预测活性值和实验活性值之间的偏差最小。
第三部分最右边的表格总结了训练集中每个化合物同某一个药效团模型的匹配信息。表格最后一栏显示了每个化合物同药效团模型的各个特征元素的匹配信息,星号*表示该特征元素未被匹配上。
一般评价药效团模型的通用规则是活性最高的两个化合物应该同模型中所有特征元素都匹配上。在该例中,只有模型5、7、8满足该条件。而且基于测试集验证数据,模型5、8也是其中两个验证数据较好的模型[测试集验证统计数据及测试集LogEstimate vs LogActiv的相关性曲线(图4)]。因此接下去可以重点关注这两个模型进行后续研究。当然该规则并非必需条件,具体情况具体分析,例如活性最高的化合物虽然没有匹配上全部的特征元素但是能匹配上的特征元素匹配的更好,或者对于某些比较感兴趣的化合物相关性比较好等等。
基于药效团模型进行化合物的设计改造
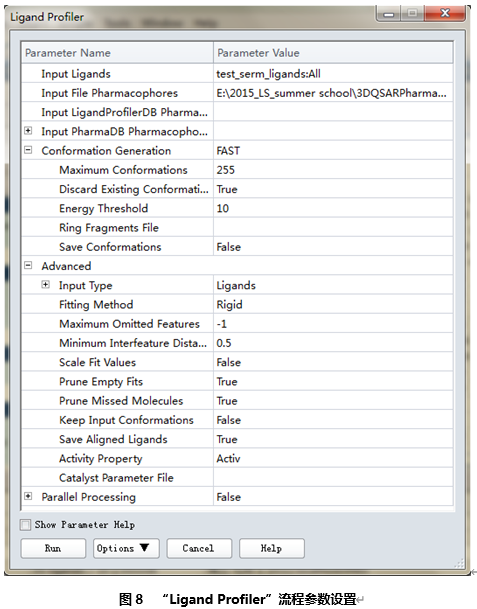
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Search, Screen and Profile,点击Ligand Profiler。
流程对应参数在参数浏览器中打开。
该流程可以将多个分子同多个药效团模型快速匹配。
设置Input Ligands为test_ serm_ligands:All。
点击Input File Pharmacophores右下拉菜单,选择Browse选项,找到之前构建药效团模型所得到的output文件。
CTRL-选取药效团模型5、8(或者SHIFT-选取所有模型)。
展开Conformation Generation参数组,点击Conformation Generation右边的栅格,下拉列表中选择FAST,能量阈值Energy Threshold设为10。
展开Advanced参数组,将Maximum Omitted Features右边的数值改为-1以允许部分匹配。点击Scale Fit Values右边的栅格,下拉列表中选择False。
点击Save Aligned Ligands右边的栅格,下拉列表中选择True。
其余参数使用默认值。(图8)
点击Run运行作业。
点击Background等待作业完成。
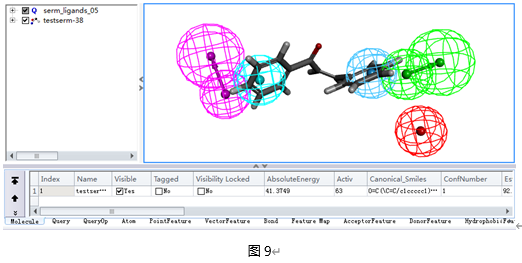
作业完成后,展开作业浏览器(Jobs Explorer)中相应的行,点击serm_ligands_05_view.pl查看测试集分子同模型5的匹配情况。
结果显示了29个测试集分子中每个分子同模型5匹配得到的最高FitValue(最低Estimate)的构象。
勾选testserm-38化合物的visible复选框,CTRL-选取化合物testserm-38和药效团模型serm_ligands_05,复制粘贴到新的分子窗口。(图9)
该化合物的实验活性值为63μM,预测活性值为93μM。该化合物只能同该模型五个特征元素中的两个特征元素匹配上。该匹配构象的能量值比较低,因此不足为虑。在有些情况下,该值会比较高,应具体分析。
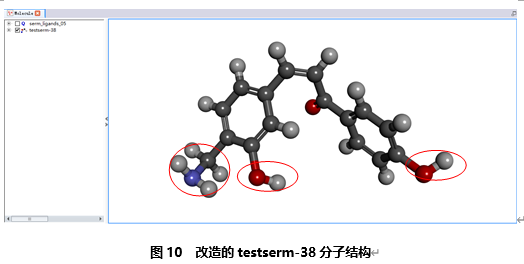
针对该化合物未能匹配的药效团特征元素,可以对该分子在结构上作两个小的改造。
在系统视图(Hierarchy View)中,关闭serm_ligands_05药效团模型的复选框。
对testserm-38进行结构改造,在苯醛基的苯环对位加一个酚羟基,在另一个苯环间位加一个酚羟基,对位加一个亚甲基胺。
修改之后的分子如图10所示:
在系统视图(Hierarchy View)中,重新选取serm_ligands_05药效团模型的复选框。
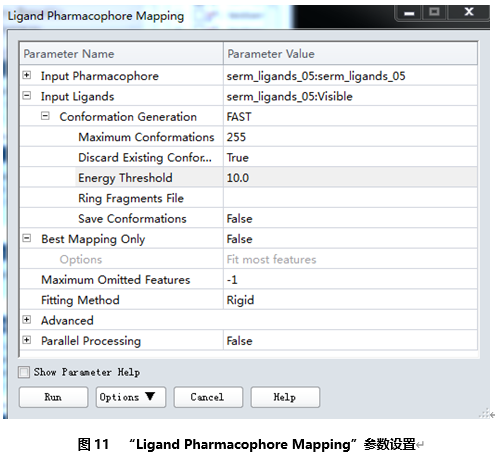
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Search, Screen and Profile,点击Ligand Pharmacophore Mapping。
流程对应参数在参数浏览器中打开。
设置Input Ligands为当前窗口中改造过后的testserm-38分子。
设置Input Pharmacophore为当前窗口中的serm_ligands_05药效团模型。
点击Best Mapping Only右边的栅格,下拉列表中选择False。设置Maximum Omitted Features为-1。
展开Input Ligands/ Conformation Generation参数组,能量阈值Energy Threshold设为10,其他参数均使用默认值。(图11)
点击Run运行作业。
等待作业完成。该作业几秒钟即可完成。
双击作业浏览器(Jobs Explorer)窗口中相应的行,打开Report页面点击ViewResults按钮。
在表格浏览器中点击 键,查看改造之后的testserm-38预测活性最高的匹配构象。
键,查看改造之后的testserm-38预测活性最高的匹配构象。
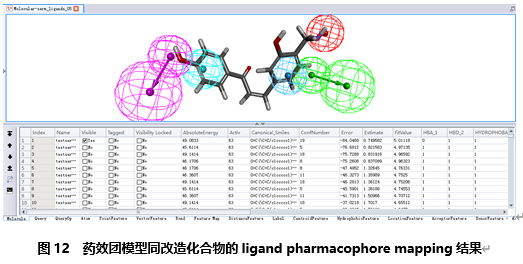
结果显示改造之后的化合物预测活性小于1μM。药效团模型五个特征元素的四个都被匹配上(图12),构象能量值仍保持较低的值。
在表格浏览器中点击 键,查看该化合物第五个匹配构象。
键,查看该化合物第五个匹配构象。
结果显示匹配情况有所改变。这一现象说明对不同的匹配情况进行研究有可能可以发现新的结合模式。
双击Pharmtype一栏两次,使匹配的特征元素个数由高到低排列,单击![]() 按钮。
按钮。
可以发现药效团模型五个特征元素都被匹配上,该匹配构象的预测活性值为3.20μM。因此在有些情况下,化合物与特征元素匹配度更高比匹配的特征元素多但匹配度低要更好。
提示:如果运行某一个流程时经常使用相同的设置,但又不想在每次操作时对参数重新进行设定,可以在设定完参数以后,在参数设置浏览器中点击Options,选取Save Protocol Settings以保存参数设置。

https://m.sciencenet.cn/blog-3536821-1362022.html
上一篇:Discovery Studio官方教程(Help-Tutorials) 基于MODELER构建抗体模型
下一篇:Discovery Studio官方教程(Help-Tutorials) 构建基于分子共同特征的药效团模型