博文
代谢学人——Nature Metabolism:线粒体:我裂开了
||
代谢学人
Nature Metabolism:线粒体:我裂开了
撰文 | 郑宇含 张彦康 张婷 仲银召 黄子晗 李雨
编辑 | 孟美瑶
校对 | 张婷

研究背景
肥胖已成为世界性流行病,它极大地增加了2型糖尿病、非酒精性脂肪性肝炎等代谢相关疾病的发病率。在肥胖发展过程中,白色脂肪组织(WAT)长期扩张并经历以激素不敏感、炎症、纤维化和细胞凋亡为特征的代谢变化。其中,线粒体在脂肪细胞代谢稳态中发挥着重要作用,例如在脂肪产热过程中线粒体通过氧化燃料以产生ATP和热量,但在肥胖患者的脂肪组织中线粒体功能受损。然而线粒体功能受损的机制,以及线粒体功能受损如何引起肥胖及其相关代谢疾病的,仍未可知。
值得注意的是,线粒体功能障碍是人类和啮齿类动物肥胖、胰岛素抵抗和脂肪肝疾病的特征。与健康个体相比,肥胖个体肌肉和脂肪组织中线粒体氧化功能减弱,并伴随脂肪细胞中线粒体含量减少及肌肉中线粒体碎片化。线粒体功能、数量及形态的变化受线粒体动力学即线粒体融合与分裂调控。其中融合过程主要调控线粒体数量和完整性,主要受Opa1调控;而分裂主要在细胞分裂过程中介导线粒体的分裂和质量控制过程(小编注:线粒体网络结构的动态变化取决于线粒体融合和分裂之间的平衡,线粒体不断的进行融合和分裂,从而维持细胞生存状态和保持线粒体的形状、大小和数量,这对线粒体代谢功能有重要影响。一方面,线粒体融合确保了细胞中保持所需数量的健康线粒体;另一方面,线粒体分裂是为了确保受损线粒体的移除。故而分裂是为了移除受损线粒体,融合是为了维持健康线粒体数量及其完整性。同时,分裂/融合是一个动态平衡过程;如出现分裂异常会导致线粒体破碎,而融合异常则会导致线粒体形态延长,两者都会影响线粒体的功能,从而导致相关疾病的发生),主要受Drp1调控。然而,在许多非分裂细胞中也会观察到线粒体的融合和分裂现象,这表明这一过程的动态平衡对于细胞适应性能量需求是至关重要的。
Ral GTPase是Ras超家族的成员,参与多种细胞过程,包括胞吐作用、增殖和自噬等,其GTP酶活性受上游调节因子Ral-GTPase活化蛋白(RalGAP)复合物调控。本文课题组前期研究表明,胰岛素通过抑制RalGAP复合物的磷酸化以及RGL2(RGL2是RalA的鸟嘌呤核苷酸交换因子)的定位激活脂肪细胞RalA,随后RalA通过与囊泡复合体的相互作用,将GLUT4囊泡转移至质膜并融合,进而促进脂肪细胞葡萄糖摄取(小编注:Ral GTPases是一类由RAS相关基因表达的蛋白,包括两种高度同源的Ral-GTPase:RalA和RalB。Ral GTPases在GTP结合的活性状态和非活性状态之间不断变化,活性状态的Ral GTPases可与一系列效应蛋白结合,从而参与多个细胞过程的信号通路,对细胞生长、分化、存活、增殖等多种功能的调节发挥重要作用)。此外,脂肪组织特异性敲除RalGAPB(RalGAP复合物中的支架蛋白亚基)可激活RalA,促进BAT葡萄糖摄取,进而改善HFD小鼠的葡萄糖稳态。同样,骨骼肌特异性敲除RalGAPA1(RalGAP复合物中的催化亚基)也可以改善小鼠的机体糖稳态(小编注:本文课题组之前的系列研究发现,胰岛素−PKB通路可磷酸化RalGAPα1-Thr735位点,从而抑制RalGAPα1活性,进而激活小G蛋白RalA,而RalA的活化会促进葡萄糖转运体GLUT4和脂肪酸转运蛋白CD36从细胞浆向细胞膜移位,从而介导骨骼肌细胞对葡萄糖和脂肪酸的摄取。在RalGAPα1 T735A基因敲入小鼠中,胰岛素无法磷酸化RalGAPα1,导致骨骼肌细胞对葡萄糖和脂肪酸的摄取受阻,最终导致突变小鼠出现肥胖、高血糖、高血脂、葡萄糖不耐受和胰岛素抵抗等代谢病症。另外,脂肪特异性敲除RalGAPB能激活RalA,主要促进BAT的葡萄糖摄取,抑制iWAT的葡萄糖摄取,整体上改善小鼠葡萄糖处理能力)。
在本篇研究中,研究人员发现肥胖小鼠白色脂肪组织中RalA基因和蛋白表达及其活性增加,并且白色脂肪组织中RalA缺失可增加机体能量消耗和线粒体氧化磷酸化水平,进而改善HFD诱导的肥胖。而白色脂肪组织RalA缺失所带来的代谢益处主要是由iWAT组织中线粒体分裂水平下降所驱动的。体外研究表明,RalA可与蛋白磷酸酶PP2Aa相互作用,促进Drp1 S637位点的去磷酸化,提高Drp1活性,进而导致线粒体过度分裂和破碎。

敲黑板啦!
1. 脂肪特异性敲除RalA可改善HFD小鼠肥胖、肝脏脂肪变性和糖稳态;
2. 脂肪细胞中敲除RalA促进线粒体活性和脂肪酸氧化;
3. 靶向敲除RalA可防止iWAT中肥胖诱导的线粒体分裂;
4. RalA促进Drp1 Ser637去磷酸化,促进脂肪细胞线粒体分裂。
研究结果
1.白色脂肪细胞特异性缺失RalA有助于小鼠抵抗HFD诱导的肥胖
研究人员提取高脂(HFD)和普通饮食(CD)小鼠脂肪的成熟脂肪细胞进行RNA-seq,结果发现与CD小鼠相比,在HFD小鼠eWAT和iWAT组织RalA基因表达显著上调,而Ralgapa2基因表达显著下降(图1a-b);此外,HFD小鼠iWAT组织RalA蛋白水平和活性(RalA-GTP结合)显著上调,而eWAT组织中RalA蛋白水平下降,活性没有明显变化(图1c-d,附图1a-b),但BAT中RalA表达水平无明显变化(附图1c)。总之,这些结果表明肥胖小鼠白色脂肪细胞中RalA活性升高。
为了进一步探究RalA是否在机体葡萄糖稳态和能量代谢中发挥作用,研究人员构建了脂肪特异性敲除RalA的小鼠(RalAAKO),其iWAT原代脂肪细胞、和BAT中RalA蛋白水平降低了90%以上,在eWAT和肝脏中RalA蛋白水平降低了约50%(小编注:本文使用了脂肪特异性敲除RalA的小鼠,研究人员提取了iWAT中成熟脂肪细胞,可能因为没有其他类型细胞表达RalA干扰蛋白水平的检测,检测发现其敲减效率较高,而组织中存在其他类型的细胞表达RalA,所以eWAT和肝脏组织中检测RalA蛋白水平较高,体现为敲除效率较低。文献显示,在脂肪组织中,RalA在巨噬细胞、成纤维细胞和雪旺细胞中表达较高,推测eWAT组织中上述细胞类型所占比例较多从而导致eWAT组织整体敲减效率比BAT低),而RalA蛋白水平在肝脏中没有变化(附图1d)。与对照组相比,胰岛素刺激下RalAAKO小鼠的WAT和原代脂肪细胞中RalA活性下降(附图1e)。脂肪特异性敲除RalA仅在iWAT中抑制了胰岛素刺激的葡萄糖摄取(附图1f-h)。但先前的研究表明RalA在调节BAT的葡萄糖摄取中也起重要作用,因此研究人员将RalA-floxed与UCP1-cre转基因小鼠杂交构建了棕色脂肪特异性敲除RalA的小鼠(RalABKO)(附图1i),发现在BAT中特异性敲除RalA减少了BAT的葡萄糖摄取(附图1j-l)。此外,RalA在两种敲除模型中对eWAT的葡萄糖摄取均无明显影响。总之,这些结果说明在RalAAKO小鼠中,RalA主要抑制iWAT中的葡萄糖摄取。接下来,为了进一步探究RalA是否以细胞自主的方式调控脂肪细胞摄取葡萄糖,研究人员利用iWAT的原代脂肪细胞,发现敲除RalA完全抑制了胰岛素诱导的GLUT4质膜易位及葡萄糖摄取,但不影响上游的胰岛素信号转导(附图1m-p)(小编注:本文作者之前的PNAS文献中提到,研究人员在BAT中用胶原酶消化分离脂肪细胞,洗涤,然后在蔗糖缓冲液中将脂肪细胞匀浆,并在蔗糖缓冲液中进行密度梯度离心,分离出高密度微粒体、低密度微粒体和质膜。图中KO原代细胞中RalA没有敲干净可能是因为此处提取的是iWAT中的SVF细胞并诱导分化后提取质膜,除了成熟脂肪细胞,可能混有部分成纤维细胞,成纤维细胞中RalA表达量较高,所以在KO细胞中能检测到RalA蛋白表达,与此对应的是附图1d中研究人员直接提取iWAT的成熟脂肪细胞检测RalA的表达,结果显示敲除得较干净。另外,从图上看WT KO加胰岛素处理后质膜组分GLUT4水平差异并不是很大,同样的糖摄取的抑制程度也不高。此前该课题组2007年发表在Dev Cell的文章中就表明胰岛素激活RalA,促进GLUT4囊泡与质膜的对接从而促进GLUT4质膜易位,而敲减/突变RalA会抑制GLUT4的质膜易位从而抑制糖摄取,并且此前发表在PNAS上的文章也表明抑制RalA会显著抑制脂肪细胞中GLUT4膜易位和葡萄糖摄取;本文中KO细胞中质膜组分的GLUT4水平和糖摄取没有非常受影响可能是存在其他调控GLUT4质膜转移的机制以及其他影响糖摄取的机制),说明RalA以细胞自主方式发挥作用。
在CD条件下,尽管RalAAKO小鼠的体重与对照组相似,但RalAAKO小鼠体脂和三种脂肪组织重量都显著下降(附图2a-c),组织学分析发现iWAT脂肪变小,但eWAT和BAT无明显差异(附图2d)。此外,RalAAKO小鼠葡萄糖稳态没有明显差异,而胰岛素敏感性略有下降(附图2e-f),并且RalAAKO小鼠胰岛素水平和胰岛素抵抗稳态评估(HOMA-IR)(小编注:测量过夜禁食小鼠的葡萄糖和胰岛素水平,HOMA-IR=空腹胰岛素水平(μU/mL) ×空腹血糖(mmol/L)/22.5)与对照组相比无明显变化(附图2g-h)。接下来,研究人员对RalAAKO小鼠进行HFD喂养,发现在HFD喂养下,与对照组相比,RalAAKO小鼠体重较轻、体脂较少(图1e-f),但三种脂肪中只有iWAT重量显著下降(图1g)。与CD相同,HFD喂养的RalAAKO小鼠iWAT脂肪细胞变小,eWAT和BAT无明显差异(附图2d)。尽管RalAAKO和对照小鼠的空腹血糖在CD和HFD下均无明显差异(附图2i-j),但HFD喂养的RalAAKO小鼠葡萄糖稳态显著改善,胰岛素敏感性没有明显变化,但胰岛素水平降低,HOMA-IR水平下降(图1h-k)。
鉴于上文发现RalAAKO在HFD条件下可能是通过减少iWAT重量减轻小鼠体重,为进一步确认RalA是否主要在WAT而不是BAT中发挥作用,研究人员监测了RalABKO小鼠的代谢表型,发现在CD喂养的RalABKO小鼠中,体脂、iWAT、eWAT重量等与WT相比均无明显差异,但BAT重量减少,推测可能由于BAT葡萄糖摄取减少,尽管RalABKO小鼠的葡萄糖稳态和胰岛素敏感性无明显变化(附图2k-n)。在HFD喂养的RalABKO小鼠中,体重、体脂、组织重量、GTT、ITT也无明显差异(附图2o-s),但研究人员也发现HFD喂养的RalABKO小鼠在BAT中表现出胰岛素抵抗(本文未给出数据也未作说明。)。这些结果说明,在BAT中敲除RalA对小鼠体重无明显影响,因此HFD喂养的RalAAKO小鼠体重显著下降主要与WAT(特别是iWAT)中RalA的敲除相关。
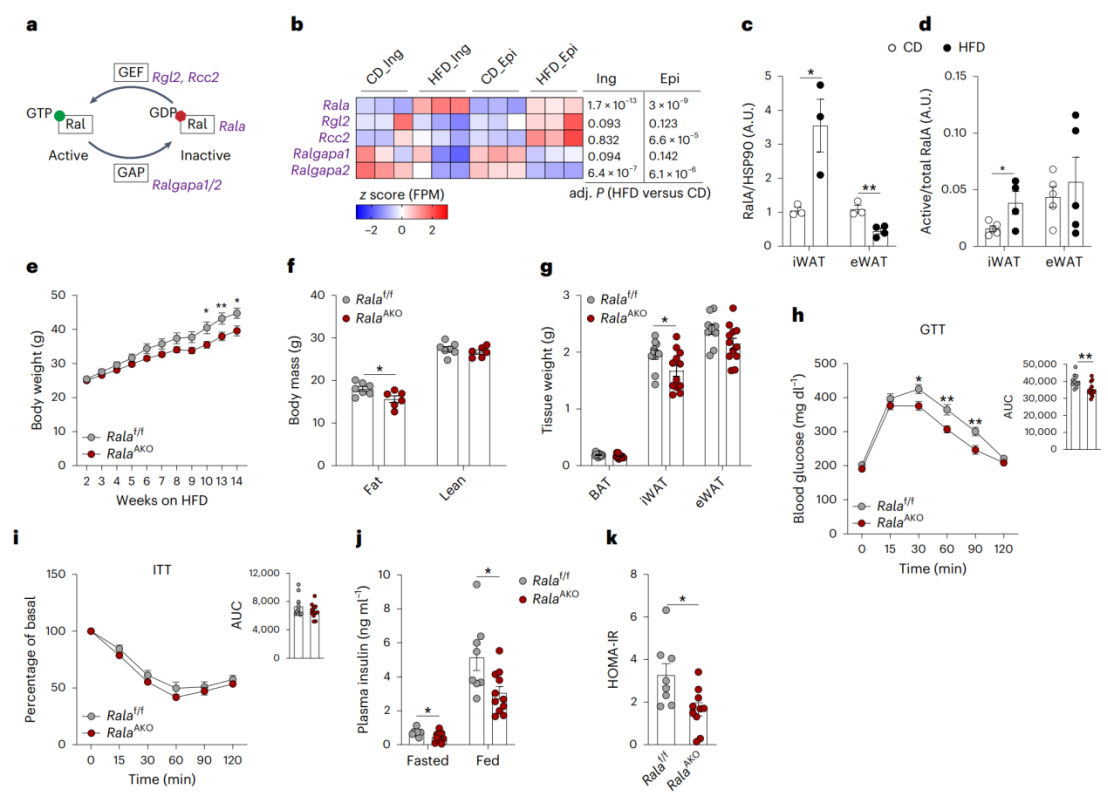
图1.WAT特异性缺失RalA有助于HFD小鼠抵抗肥胖
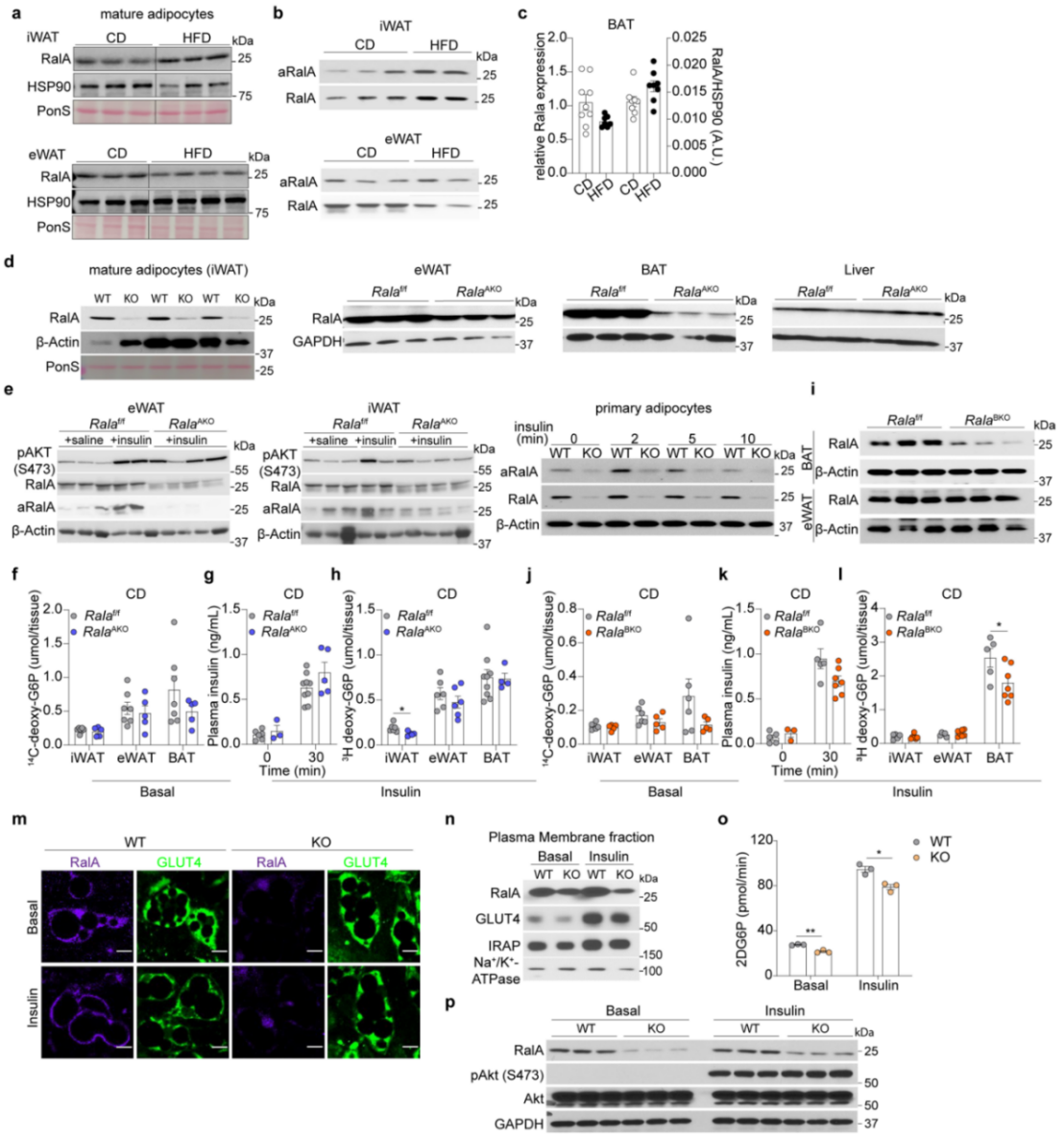
附图1.肥胖脂肪细胞中RalA蛋白含量和活性增加
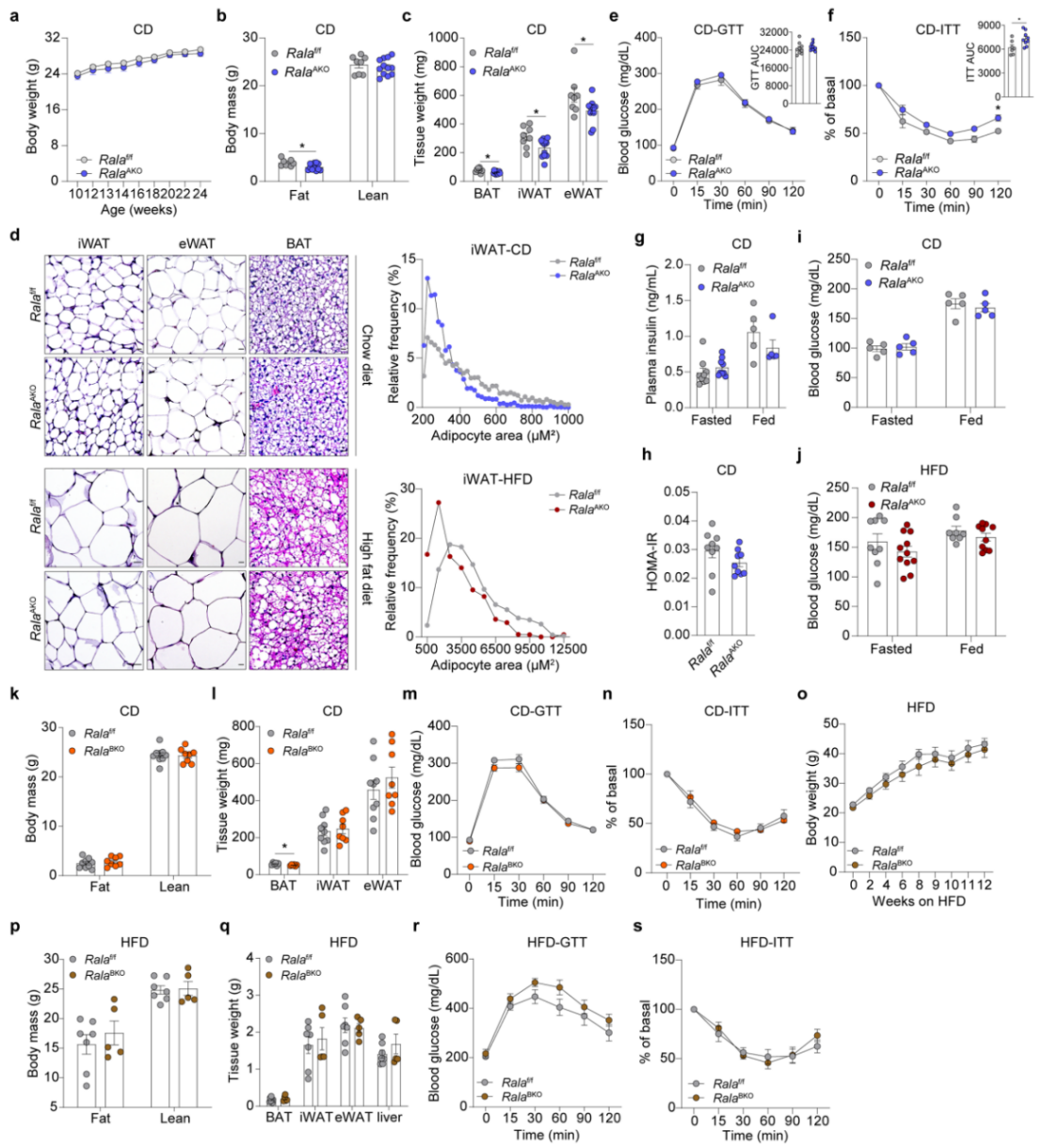
附图2.RalABKO小鼠与RalAAKO小鼠表型不相似
2.WAT缺失RalA能改善HFD诱导的肝脂肪变性
由于HFD喂养的RalAAKO小鼠GTT结果呈改善趋势而ITT结果无差异,研究人员推测葡萄糖处理能力改善是肝脏葡萄糖产生减少所致,于是研究人员检测了脂肪特异性敲除RalA对肝脏产糖能力的影响。研究人员在HFD喂养的小鼠中进行丙酮酸耐受实验(PTT),发现与对照小鼠相比,RalAAKO小鼠中丙酮酸诱导的葡萄糖产生减少(图2a)。同时,肝脏糖异生基因G6pc和Pepck表达显著下调(图2b)。总之,脂肪细胞特异性缺失RalA可通过减少肝脏葡萄糖生成来改善葡萄糖稳态。
随后,研究人员发现HFD喂养的RalAAKO小鼠肝脏重量和甘油三脂(TG)含量显著降低(图2c-d),同时H&E和油红O染色显示RalAAKO小鼠肝脏脂质积累减少(图2e),并且RalAAKO小鼠肝脏中脂肪生成基因(Acc,Fasn,Scd1和Acsl1)表达水平降低(图2f)。然而,RalAAKO小鼠血浆瘦素水平(小编注:除了常见的瘦素通过减少热量摄入诱导肥胖进而产生脂肪肝以外,目前有研究认为瘦素能独立于食物摄入改善肝脏脂质积累,例如有研究表明大脑中的瘦素信号能通过迷走神经,促进肝脏极低密度脂蛋白甘油三脂(VLDL-TG)分泌,从而减轻肝脏脂质积累(Cell Metablism,2022),而中枢神经系统瘦素通过脑-迷走神经-肝轴抑制肝脏脂质积累被认为可能是新的肥胖治疗途径。所以这里作者检测了瘦素。)和肝脏中脂肪酸氧化(FAO)相关基因的表达均没有明显差异(图2g-h)。此外,与对照组相比,RalAAKO小鼠的肝脏中炎症相关基因(Adgre1)和纤维化相关基因(Col1a1和Col3a1)的表达水平较低(图2i),天冬氨酸氨基转移酶(AST)和丙氨酸氨基转移酶(ALT)的活性也较低(图2j-k)。值得注意的是,HFD喂养的RalABKO小鼠肝脏重量与对照组相似(附图2q)。总之,这些结果表明,WAT特异性缺失RalA能系统性调节脂质代谢,从而改善肥胖诱导的肝脏脂肪变性和损伤。
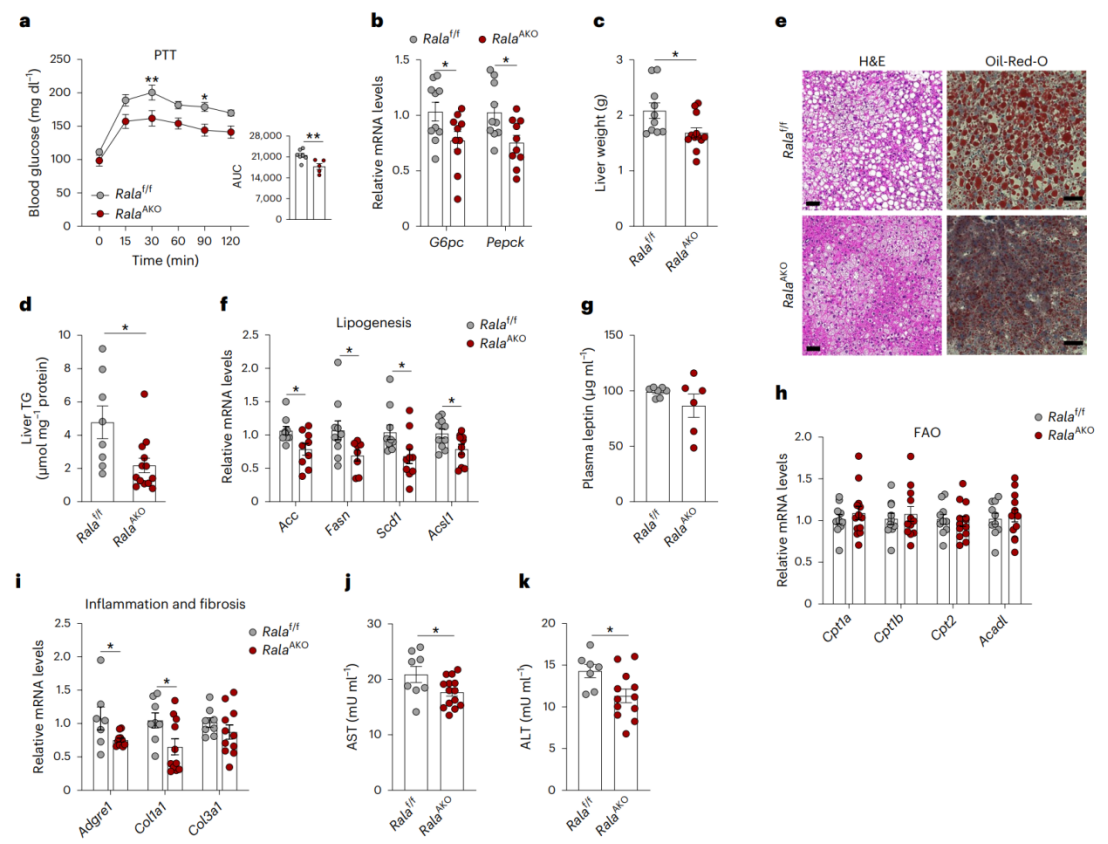
图2. WAT缺失RalA能改善HFD诱导的肝脂肪变性
3.WAT缺失RalA能促进能量消耗和线粒体氧化磷酸化
接下来,研究人员进一步探究脂肪组织缺失RalA改善HFD诱导的肝脂肪变性、肥胖和葡萄糖耐受不良的机制。研究人员发现,在CD喂养条件下,脂肪细胞缺失RalA对小鼠的能量代谢和摄食量没有影响(附图3a-e)。而HFD喂养的RalAAKO小鼠在黑暗阶段能量消耗增加(图3a),并且RalAAKO小鼠耗氧量也显著增加(附图3f),但呼吸交换率(RER)、活动量和摄食量无明显差异(附图3g-i)。此外,HFD喂养的RalABKO小鼠能量消耗、耗氧量、活动量和摄食量均与对照组相似(附图3j-n)。总之,这些结果表明,WAT特异性缺失RalA促进了机体能量消耗。
有研究表明,能量消耗的增加与线粒体氧化活性增加相关。随后,研究人员发现与对照组相比,HFD喂养的RalAAKO小鼠iWAT中氧化磷酸化(OXPHOS)相关蛋白表达显著增加(图3b-c),而在eWAT中没有明显变化(附图3o-p)。值得注意的是,之前的结果表明HFD喂养的RalABKO小鼠代谢表型没有变化,但RalAAKO小鼠BAT中复合物Ⅰ和复合物Ⅱ的水平上升(附图3q-r),因此这可能不是BAT的细胞自主性功能,而是RalAAKO小鼠全身代谢改善的影响。随后,研究人员发现HFD喂养的RalAAKO小鼠血浆游离脂肪酸(FFA)和TG水平较低(图3d-e)。最近的研究表明,iWAT棕色化能促进能量消耗并抑制饮食诱导的肥胖。于是,为了探究RalA是否影响iWAT棕色化,研究人员检测了HFD小鼠三种脂肪中产热相关基因(UCP1、Cidea、Prdm16)的表达,发现脂肪缺失RalA并不影响三种脂肪中产热基因的表达,这表明RalAAKO小鼠中能量消耗的改善与脂肪组织棕色化无关(附图3s)。
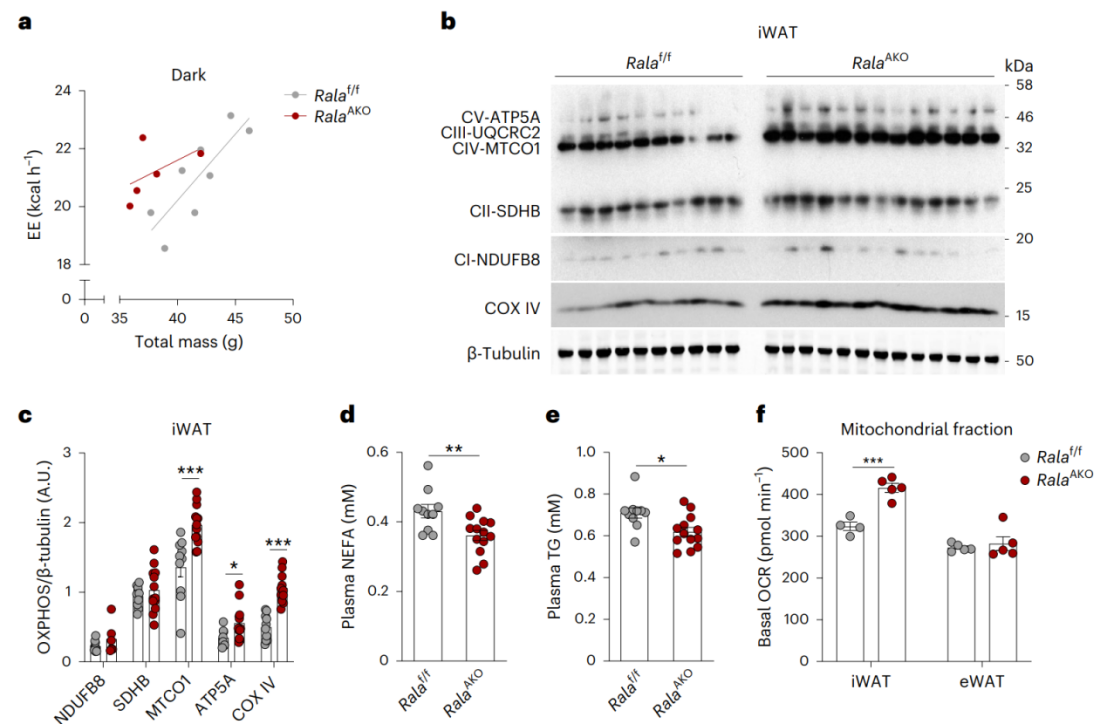
图3. WAT缺失RalA能促进能量消耗和线粒体氧化磷酸化
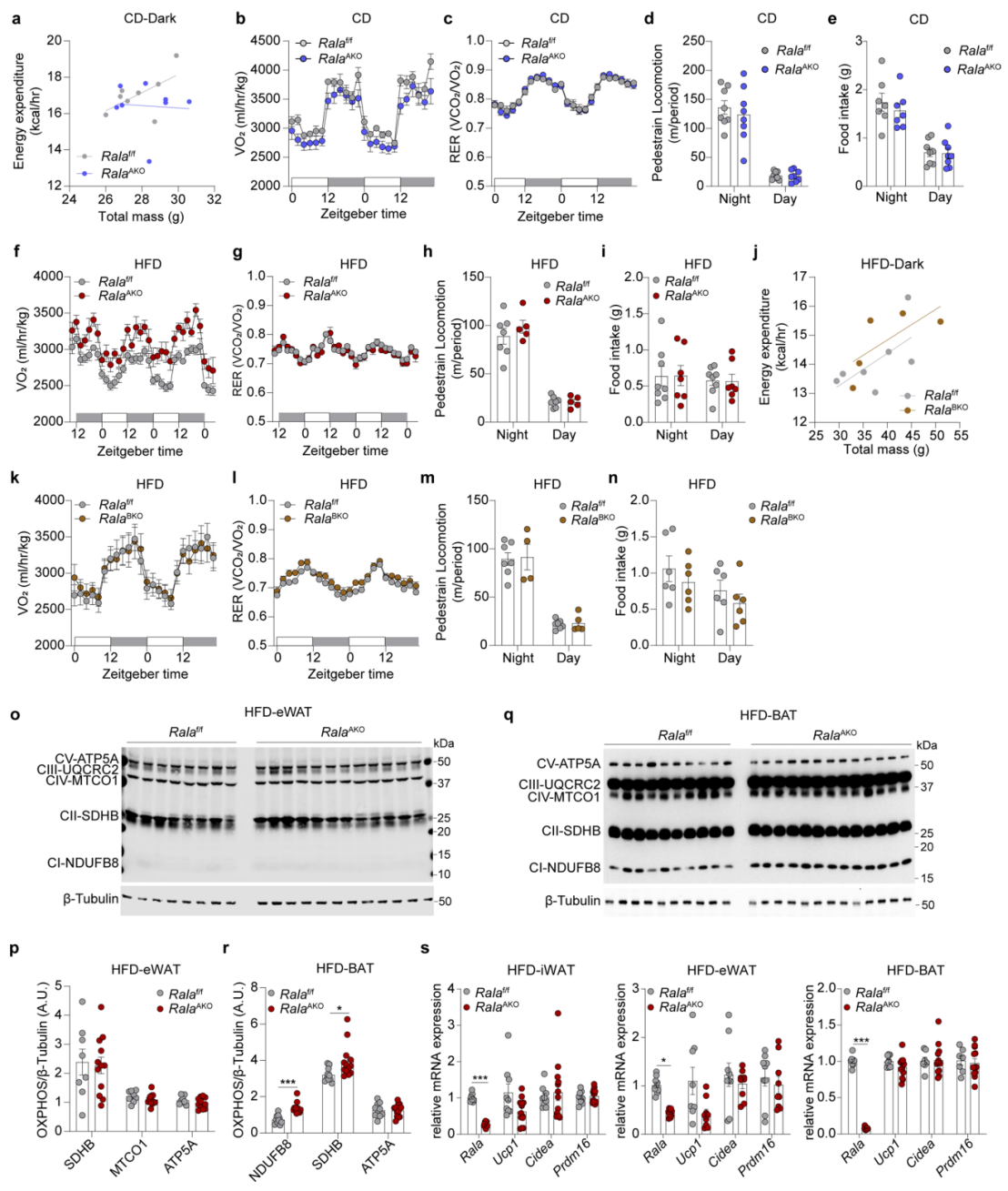
附图3. CD喂养的RalAAKO小鼠和HFD喂养的RalABKO小鼠能量消耗均没有增加
4.白色脂肪细胞中敲除RalA促进线粒体活性和脂肪酸氧化
研究人员试图进一步阐明RalAAKO小鼠能量代谢改善的机制,通过直接检测脂肪细胞中的线粒体活性,发现与对照组相比,从RalA AKO小鼠iWAT中分离的线粒体基础氧耗率增加,但从eWAT中分离的线粒体没有差异(图3f)。并且,RalA AKO小鼠的原代白色脂肪细胞相比于对照组也具有更高的基础和最大氧耗率,然而,在添加CPT1抑制剂etomoxir抑制脂肪酸氧化后,这种差异消失,提示RalA可能调控脂肪酸氧化(图4a,附图4a)。为了直接确定RalA是否调控脂肪酸氧化,研究人员利用C14标记的棕榈酸(PA)处理原代脂肪细胞,并检测了脂肪酸氧化产物酸溶性代谢物(ASM)及二氧化碳水平(小编注:本实验是利用肉碱和C14标记的棕榈酸处理脂肪细胞,然后取培养基,添加高氯酸处理,检测溶于高氯酸中的代谢物及释放出来二氧化碳中的放射性含量,可代表细胞的脂肪酸氧化能力。其基本原理是棕榈酸氧化的主要产物乙酰辅酶A及其他产物或中间体如乙酰肉碱、TCA循环中间体等可溶于高氯酸,而乙酰辅酶A也可经TCA循环后产生CO2,此外,高氯酸会将未氧化的长链脂肪酸析出,因此通过检测高氯酸处理后产物即酸溶性代谢物的放射性水平及二氧化碳的放射性水平可以反应细胞的脂肪酸氧化能力。CO2不变的原因可能是由于尽管脂肪酸氧化生成的乙酰辅酶A变多了,但进入TCA循环的差异可能较小,不足以引起CO2中放射性明显的改变,仅有少量增加趋势),发现RalA AKO小鼠的原代脂肪细胞具有更高的脂肪酸氧化能力(图4b)。这些结果说明,RalA AKO在白色脂肪中通过促进线粒体氧化活性增加了能量消耗。
为进一步确证RalA的功能,研究人员构建了来源于RalAf/f小鼠的永生化前脂肪细胞系,并利用Cre慢病毒诱导RalA敲除(附图4b)。通过BODIPY和TMRM染色(小编注:TMRM含有4个甲基基团以及一个罗丹明核心,是脂溶性阳离子染料,能够以依赖于线粒体膜电位的方式进入线粒体内部积累,当线粒体膜电位较高时,TMRM信号也较强),研究人员发现,在完全分化的永生化细胞系或原代脂肪细胞中,RalA敲除增强了TMRM信号,说明RalA敲除促进了线粒体膜电位,提示RalA敲除的白色脂肪细胞线粒体具有更强的电子传递链和氧化磷酸化活性(图4c,附图4c)。同时,研究人员利用β3受体激动剂CL-316,243诱导脂肪细胞线粒体膜去极化,发现TMRM信号在激动剂处理后迅速下降,证实了TMRM染色对活性线粒体的检测能力(小编注:之前有研究提出在β肾上腺素激动剂处理后,由于解偶联效应,脂肪细胞的线粒体膜电位会降低。本文也发现在CL-316,243处理后膜电位确实会迅速降低)(附图4d)。
先前研究报道,脂肪细胞中脂解促进线粒体氧化代谢。为确认RalA敲除是否有可能主要通过促进脂解增加脂肪细胞氧化能力,研究人员分别在体外和体内检测了RalA对脂解的影响。在永生化脂肪细胞中,研究人员发现,CL-316,243在WT和RalA KO脂肪细胞系中诱导FFA和甘油释放的能力相同,且FFA和甘油的摩尔比约为3:1(附图4e,f)。在体内,研究人员也发现了类似的结果,CL-316,243在WT和RalA AKO小鼠诱导的FFA和甘油释放相同。并且,RalAAKO并不影响胰岛素对FFA释放的抑制能力(附图4e,i)。值得注意的是,在CL-316,243处理条件下,RalA KO脂肪细胞甘油释放略有增加,而RalA AKO小鼠在CL-316,243刺激或禁食后血浆甘油水平则略有下降(附图4f,h,j)。总之,这些结果表明脂肪细胞中RalA敲除增加了线粒体氧化活性,但不影响FFA供给。
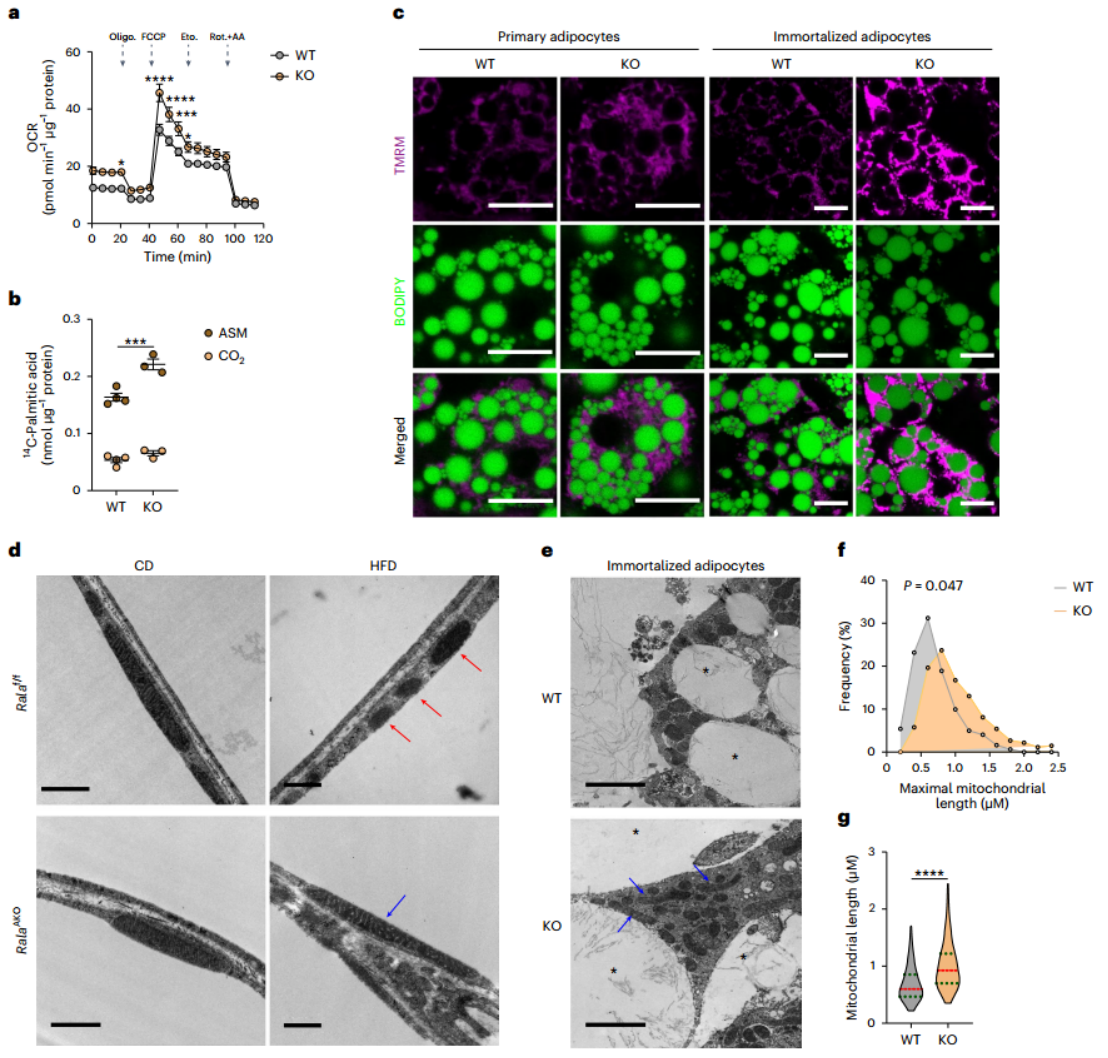
图4. 白色脂肪中RalA敲除通过抑制肥胖诱导的iWAT线粒体分裂,增强线粒体活性和脂肪酸氧化
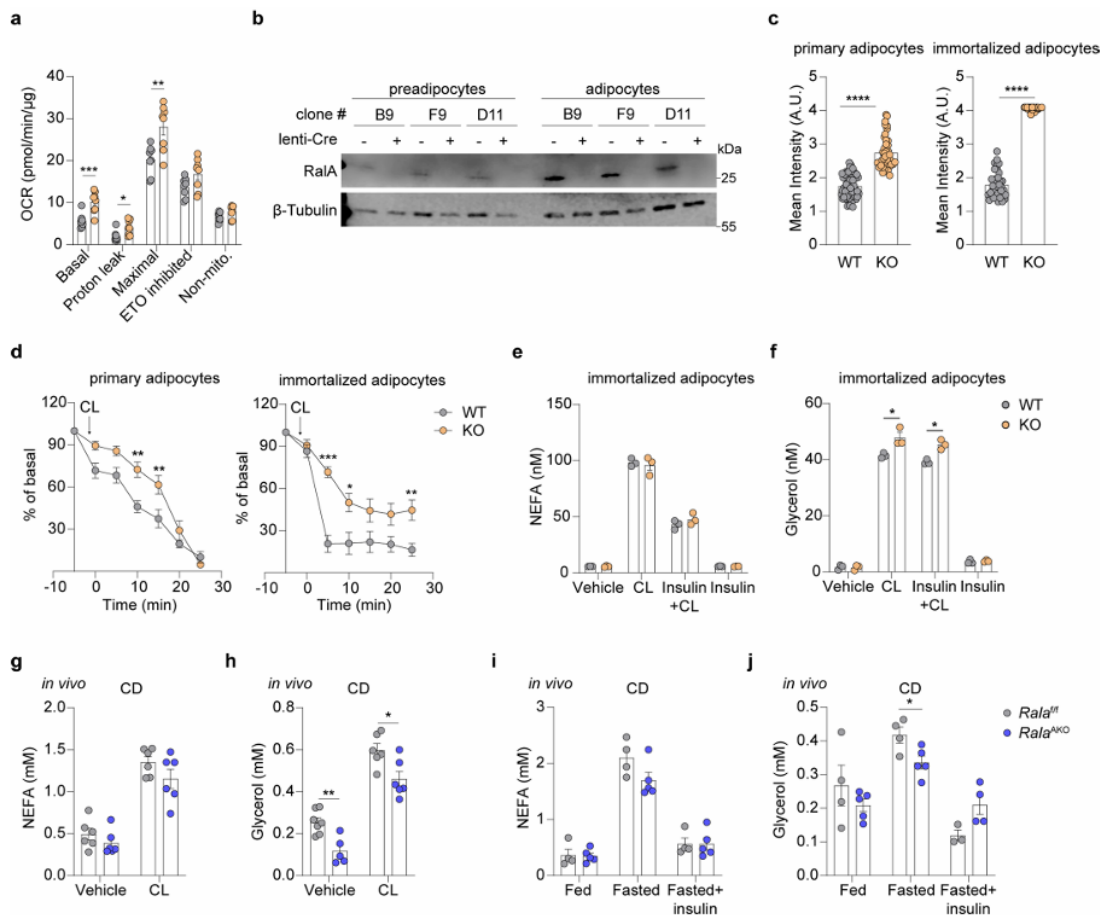
附图4. 脂肪细胞中RalA敲除不影响游离脂肪酸释放
5.靶向敲除RalA可在iWAT中防止肥胖诱导的线粒体分裂
为进一步探索RalA AKO小鼠在HFD喂养后,线粒体氧化活性增加的原因,研究人员首先检测了RalA敲除是否会影响线粒体生物发生。然而,在WT和RalAAKO的WAT中,线粒体生物发生相关基因表达并无差异(附图5a,b),且线粒体生物发生的主要调节因子AMPK活性也相同(附图5c-f)。而除了线粒体生物发生,线粒体形态的动态变化如融合和分裂等事件也同样调节线粒体功能。电镜观察发现,与先前报道的在肥胖的脂肪细胞中线粒体功能和形态受损一致,HFD导致WT小鼠的iWAT中出现较小的球形线粒体,其线粒体长度与CD相比明显变小(图4d,附图5g)。在CD条件下,RalA敲除对iWAT中线粒体的形态没有影响,这也与CD条件下体内代谢表型无差异相一致,但在HFD条件下,RalA敲除阻止了HFD诱导的线粒体形态改变(图4d,附图5g)。此外,在eWAT中,RalA敲除未能阻止HFD诱导eWAT线粒体片段化,这也与RalA AKO不影响eWAT组织重量,线粒体氧化磷酸化相关蛋白水平和线粒体OCR等相一致(图1g,3f,附图3o,3p,5h)。事实上,eWAT中的线粒体在HFD条件下并没有特别明显的片段化,可能是由于它们本身就处于片段化状态,这也与内脏脂肪细胞主要发挥合成代谢功能相吻合。此外,在CD或HFD条件下,BAT中的线粒体形态也不会由于RalA敲除而改变(附图5i)。研究人员在体外检测了iWAT永生化脂肪细胞系的线粒体形态,结果与体内相一致,RalA敲除脂肪细胞的线粒体似乎较长,且伸长态的线粒体占较大比例,平均最大线粒体长度也显著高于WT细胞(图4e,4f)。
拓展阅读:线粒体形态与细胞代谢 线粒体形态主要由线粒体融合/分裂等动力学调控。融合过程会产生伸长形态的线粒体,主要由Mfn1、Mfn2和Opa1介导,其中Mfn1和Mfn2主要介导线粒体外膜融合,而OPA1则主要介导内膜融合。分裂过程则与融合过程相反,会产生片段化线粒体,在分裂过程中由Drp1发挥核心作用。由于线粒体本身与细胞代谢和能量产生密切相关,因此,线粒体动力学与细胞能量供需平衡之间存在紧密联系。通常,细胞在营养丰富的环境中生长时,线粒体倾向于片段化,氧化磷酸化受损。而在营养缺乏的细胞中,线粒体则倾向于互相融合连接,以维持高效的氧化磷酸化和ATP产生能力。通过敲除Mfn或Opa1抑制线粒体融合会导致细胞的线粒体膜电位降低,呼吸链功能下降。此外,抑制线粒体氧化磷酸化的药物通常也与促进线粒体分裂相关,如线粒体解偶联剂CCCP或FCCP,会抑制OPA1,激活Drp1从而促进线粒体分裂。在骨骼肌中钙调磷酸酶缺失的小鼠中也发现骨骼肌线粒体伸长且呼吸链活性增加。因此,似乎线粒体的融合形态主要增强氧化磷酸化功能和ATP产生,而分裂形态则与细胞能量需求抑制相关。此外,在棕色脂肪中也已证明产热过程与线粒体动力学相关,冷暴露会导致Drp1激活,促进线粒体分裂及解偶联,增加产热但不增加ATP合成。因此在脂肪产热过程中,线粒体的片段化似乎与产热活性相关。而线粒体动力学除了调控代谢外,也参与细胞分裂过程的调控。在细胞处于G1期时,线粒体经过融合过程,形成一个广泛的、相互连接的小管网络,负责促进氧化磷酸化和ATP生成,而当细胞进入S期时,线粒体发生分裂,产生片段化的线粒体,以便在有丝分裂过程中均匀分布到子细胞中。抑制线粒体分裂会导致在有丝分裂时的线粒体分裂异常,细胞数量减少。 [1] Prashant Mishra, et al. J Cell Biol . 2016 Feb 15;212(4):379-87 [2] David C Chan. Annu Rev Pathol. 2020 Jan 24:15:235-259. [3] Jakob D Wikstrom, et al. EMBO J. 2014 Mar 3;33(5):418-36. [4] David F. Kashatus, et al. Nat Cell Biol . 2011 Aug 7;13(9):1108-15.
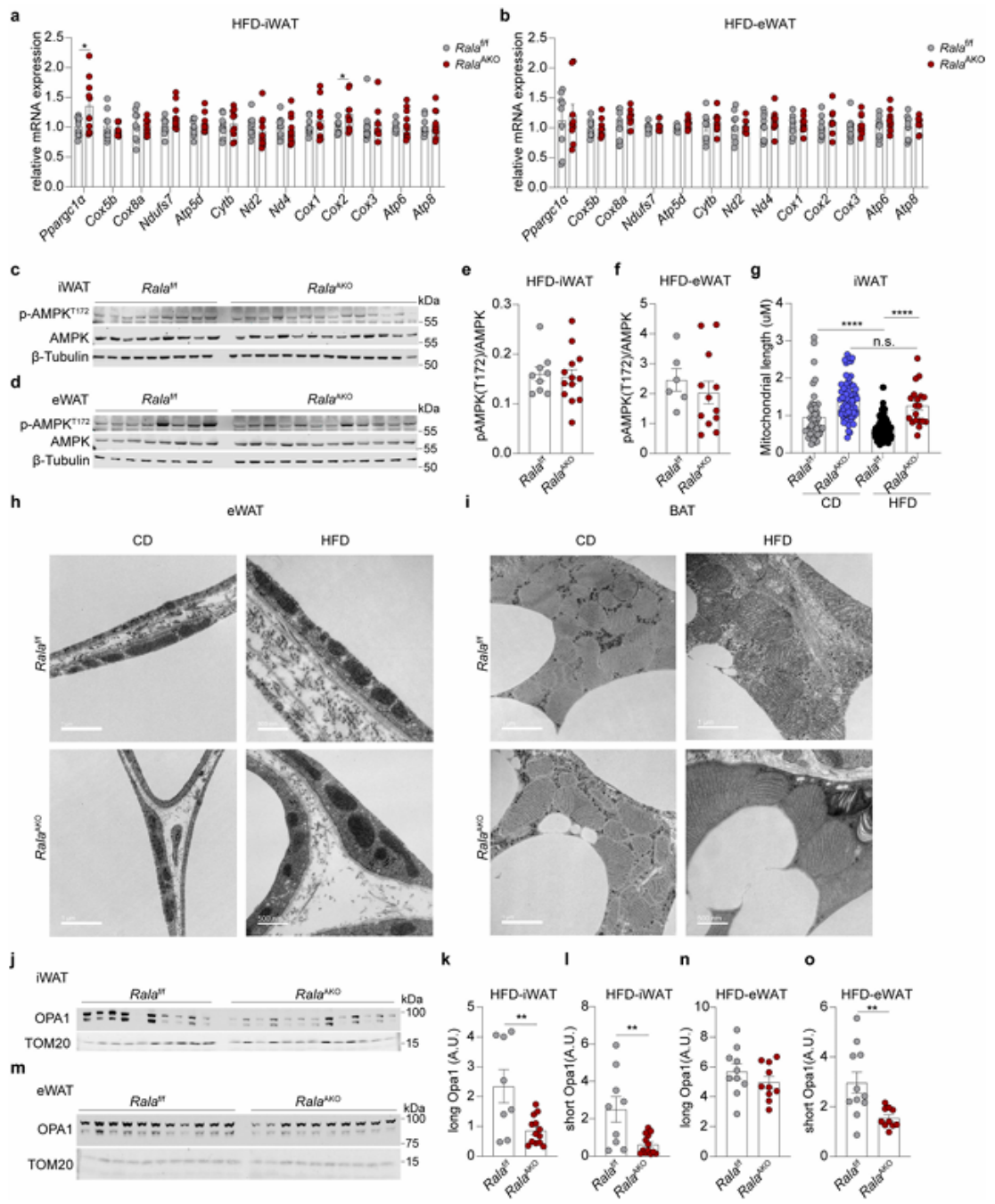
附图5. 在WAT抑制RalA不影响线粒体生物发生
6.抑制RalA在白色脂肪细胞中促进Drp1 S637磷酸化
Opa1和Drp1被认为是线粒体融合和分裂的关键调节因子。Opa1经蛋白酶切割后产生长(L-Opa1)和短(S-Opa1)两种形式,共同促进线粒体融合。在HFD小鼠的iWAT中,RalA敲除下调了这两种形式的Opa1蛋白水平(附图5j-i),在eWAT中,RalA也下调了S-Opa1的水平(附图5m-o)。这些结果提示RalA敲除可能抑制了线粒体的融合过程,但在RalA AKO小鼠的iWAT中观察到的线粒体处于伸长态,说明Opa1的表达变化可能是代偿性的过程。接下来研究人员将关注点转向促进线粒体分裂的关键调节因子Drp1(小编注:Drp1是介导线粒体分裂的主要因子,Drp1活性受不同位点磷酸化的不同调控,Drp1 S616磷酸化属于促进型磷酸化,会促进线粒体分裂,而Drp1 S637、S412和S684磷酸化都属于抑制型磷酸化,会负调Drp1功能。可能是因为S637位点比较经典,所以本文选择了S637磷酸化位点进行研究)。值得注意的是,抑制线粒体分裂相关的Drp1 S637位点的磷酸化水平在RalA敲除的iWAT中显著增加(图5a,附图6a),而eWAT中则无明显差异(附图6b,c)。β肾上腺素受体-cAMP通路通过PKA催化Drp1 S637磷酸化,为了在PKA激活的情况下评估RalA的作用,研究人员在CD喂养的小鼠中给药β3受体激动剂CL-316,243,发现RalA AKO的Drp1 S637磷酸化水平依旧更高(附图6d,e)。此外,该结果也排除了在HFD条件下,RalA AKO引起的体重差异对Drp1 S637磷酸化的间接调控。
为了确定RalA是否以细胞自主方式调控Drp1磷酸化,研究人员在永生化脂肪细胞系和原代脂肪细胞中检测RalA对Drp1磷酸化水平的影响。与体内结果一致,RalA KO脂肪细胞在Forskolin或CL-316,243处理后相比于WT具有更高的Drp1 S637磷酸化水平(图5b,c,附图6f-i)。研究人员还利用广谱Ral抑制剂RBC8(可通过将RalA保持在与GDP结合的非活性状态抑制RalA激活)预处理3T3-L1脂肪细胞,发现RBC8预处理显著促进了Forskolin诱导的Drp1 S637磷酸化(附图6j,k)。此外,在人原代脂肪细胞系SGBS中,RBC8处理也表现出一致的结果(图5d,e)。为了确定RalA是否通过调控cAMP-PKA通路发挥作用,研究人员检测了脂肪细胞中cAMP水平及PKA下游HSL的磷酸化水平,发现在CL316,243处理5分钟后,RalA敲除并不影响cAMP水平及HSL S660磷酸化水平(附图6l-n)。因此,RalA在人类和小鼠的多种脂肪细胞系中调节PKA下游的Drp1 S637磷酸化水平。
为进一步确定Drp1 S637磷酸化是否调控线粒体的氧化活性和形态,研究人员在脂肪细胞中表达Drp1 S637模拟磷酸化突变体(SD,丝氨酸突变为天冬氨酸)和模拟去磷酸化突变体(SA,丝氨酸突变为丙氨酸),发现表达Drp1SD的细胞比表达Drp1WT和Drp1SA的细胞具有更高的脂肪酸氧化能力和线粒体长度(图5f,g,附图6o),说明Drp1 S637磷酸化状态足以调控线粒体氧化能力和形态。
为探究Drp1与人类肥胖代谢调节的相关性,研究人员分析了肥胖或非肥胖女性腹部皮下WAT的微阵列数据。在人类皮下WAT中,编码Drp1蛋白的DNM1L表达水平与BMI和HOMA-IR正相关(图5h,i),并且其表达在肥胖个体中显著上调(图5j),因此推测DNM1L表达增加可能也在人类肥胖症中导致线粒体功能紊乱。并且,对来自770名男性皮下脂肪的已发表数据进行生信分析也发现DNM1L与肥胖相关(附图6p-r)。
总之,这些体内体外数据表明,脂肪组织中Drp1活性上调可能是在肥胖中导致线粒体功能障碍的重要因素,而RalA缺乏则通过促进Drp1 S637磷酸化改善了线粒体的过度分裂。
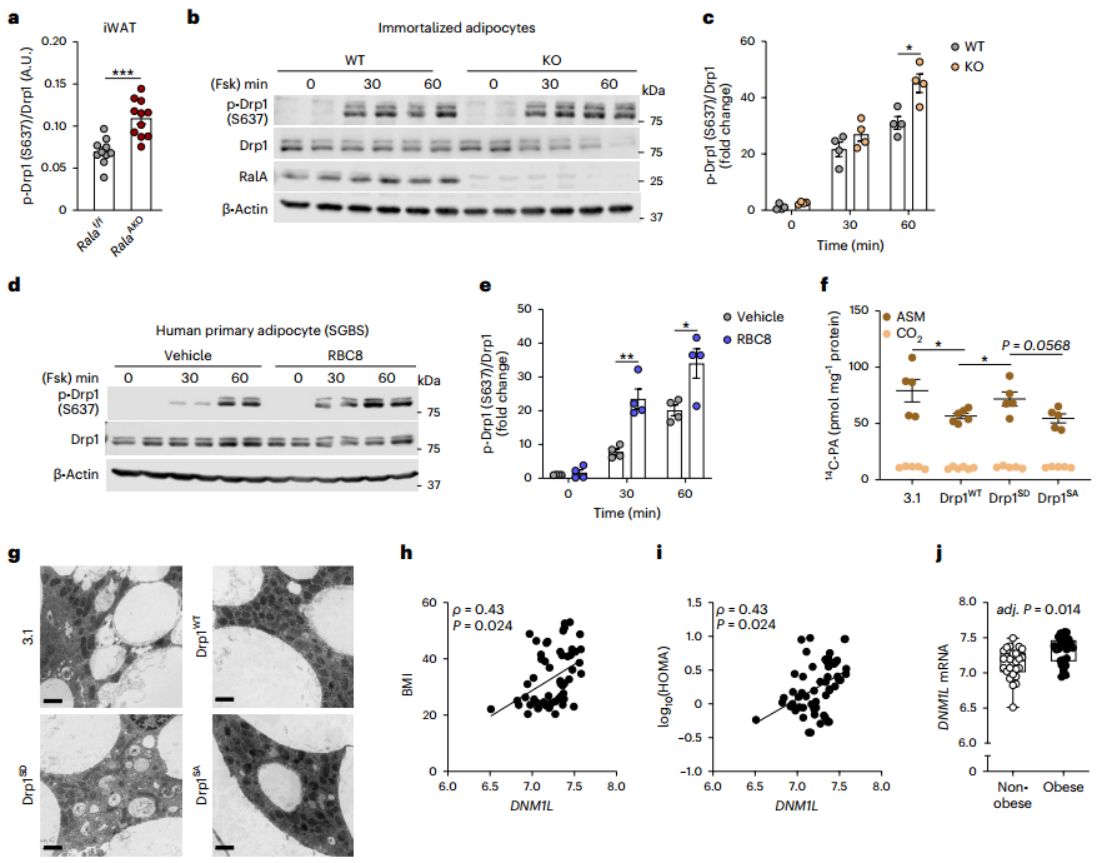
图5. 在白色脂肪细胞中抑制RalA增加了Drp1 S637位点磷酸化水平
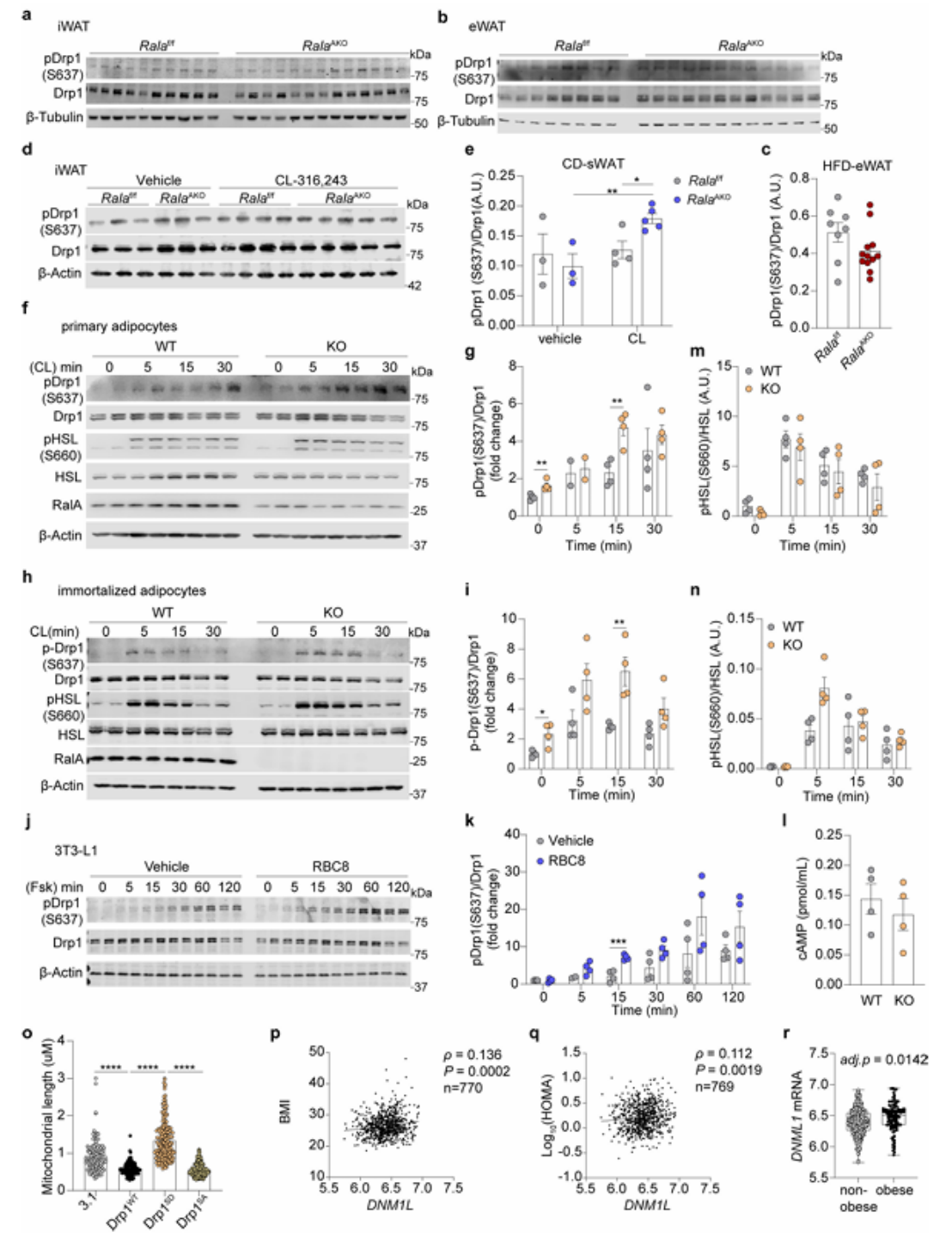
附图6. RalA敲除不影响脂肪细胞cAMP和HSL的磷酸化水平
7.RalA与Drp1和蛋白磷酸酶2A相互作用,促进Drp1 S637位点的去磷酸化修饰
为了探究RalA调控Drp1 S637位点磷酸化的分子机制,研究人员通过蛋白质组学分析来寻找与RalA WT、RalA G23V(活性激活形式突变体)、RalA S28N(活性失活形式突变体)相互作用的蛋白。在鉴定出来的结合蛋白中,蛋白磷酸酶2A亚基Aα(PP2Aa,Ppp2r1α基因编码的支架亚基)优先与RalA G23V突变体相互结合。为了证明这一结果,研究人员在HEK 239T细胞中纯化RalA WT-Flag蛋白,发现RalA WT与PP2Aa结合(图6a)。随后,研究人员在HEK 293T细胞中共表达RalA WT和RalA突变体,Sec5作为阳性对照仅与RalA G23V相互作用,结果显示,PP2Aa可与RalA WT蛋白和RalA G23V突变体结合(图6a-b)。随后,研究人员在体外用GTPγS或GDP加载RalA-Flag融合蛋白,以评估RalA与PP2Aa结合的特异性,结果显示,GTPγS负载的RalA-Flag蛋白与PP2Aa的结合作用下降,而GDP不影响RalA-Flag蛋白与PP2Aa的结合作用(图6c)。接下来,为了探究RalA是否通过PP2Aa调控Drp1磷酸化水平,研究人员在HEK 293T细胞中共表达Drp1和RalA WT、RalA G23V和RalA S28N,结果显示Drp1与RalA WT和突变体均相互作用(附图7a),而添加Forskolin激活cAMP-PKA轴,显著提高了Drp1 S637位点磷酸化,共表达PP2Aα后降低了Drp1 S637位点的磷酸化水平(图6d),但共表达PP2Ab并不影响Drp1 S637位点磷酸化水平(附图7b)。总之,这些结果表明,RalA与Drp1的相互作用关系独立于cAMP-PKA轴,激活cAMP-PKA后,RalA可通过招募PP2Aa促进Drp1 S637的去磷酸化修饰。
在WT脂肪细胞中,Drp1与RalA共定位,而在RalA KO脂肪细胞中,并未观察到这一现象(图6e,附图7c)。为了进一步探究RalA激活状态对Drp1蛋白磷酸化水平和线粒体功能的影响,研究人员在RalA KO脂肪细胞系分化前转染RalA WT和RalA G23V,结果显示,表达RalA G23V显著降低了Drp1 S637位点磷酸化水平(图6g,附图7d)。此外,表达RalA WT或RalA G23V均可显著降低RalA KO脂肪细胞的线粒体膜电位(图6h,附图7e)。为了证明线粒体膜电位降低是否与线粒体氧化功能的下降有关,研究人员对表达RalA WT和RalA G23V的Ral KO脂肪细胞进行Seahorse实验,结果显示,与RalA KO脂肪细胞相比,表达RalA WT和RalA G23V显著降低了脂肪细胞的基础OCR和最大OCR水平(图6i和附图7f)。此外EM结果显示与RalA KO脂肪细胞相比,表达RalA WT或RalA G23V促使线粒体分裂(图6j和附图7g)。活细胞成像分析也表明,与WT脂肪细胞相比,RalA KO脂肪细胞线粒体分裂水平较低,而在线粒体融合方面没有明显差异(附图7h)(小编注:这一结果是研究人员利用定制点状光片显微镜进行4D线粒体活细胞成像分析所得到的,其中488nm和560nm激光分别激发BODIPY和MitoTracker Red。具体而言,研究人员通过细胞分割、线粒体分割和线粒体运动跟踪来量化线粒体的融合和分裂,首先研究人员利用imageJ和Python脚本将不同时间内的细胞切割成单细胞,然后利用MitoGraph对每个细胞中的线粒体进行切割,随后使用MitoTNT来跟踪线粒体的运动,进行进一步的融合和分裂动力学分析)。
先前研究表明,RalA可促进增殖细胞分裂,而敲除RalA基因会抑制线粒体分裂,形成一个长而相互连接的线粒体网络,进而抑制细胞增殖水平。与本项研究一致的是,本研究发现RalA缺失导致脂肪细胞线粒体拉长,OXPHOS水平增加,进而影响全身脂质代谢;而与本项研究不同的是,本研究并没有观察到RalBP1与Drp1之间的相互作用。值得注意的是,与WT小鼠iWAT组织相比,RalA AKO小鼠iWAT组织中PP2Aa蛋白水平显著升高,而PP2Ab和PP2Ac蛋白水平无明显差异(附图7j-k)。综上所述,这些结果表明,肥胖促进RalA表达,并与PP2Aa相互结合,进而促进Drp1 S637位点发生去磷酸化修饰,而这一现象导致肥胖小鼠脂肪细胞中线粒体发生碎片化(附图8)。
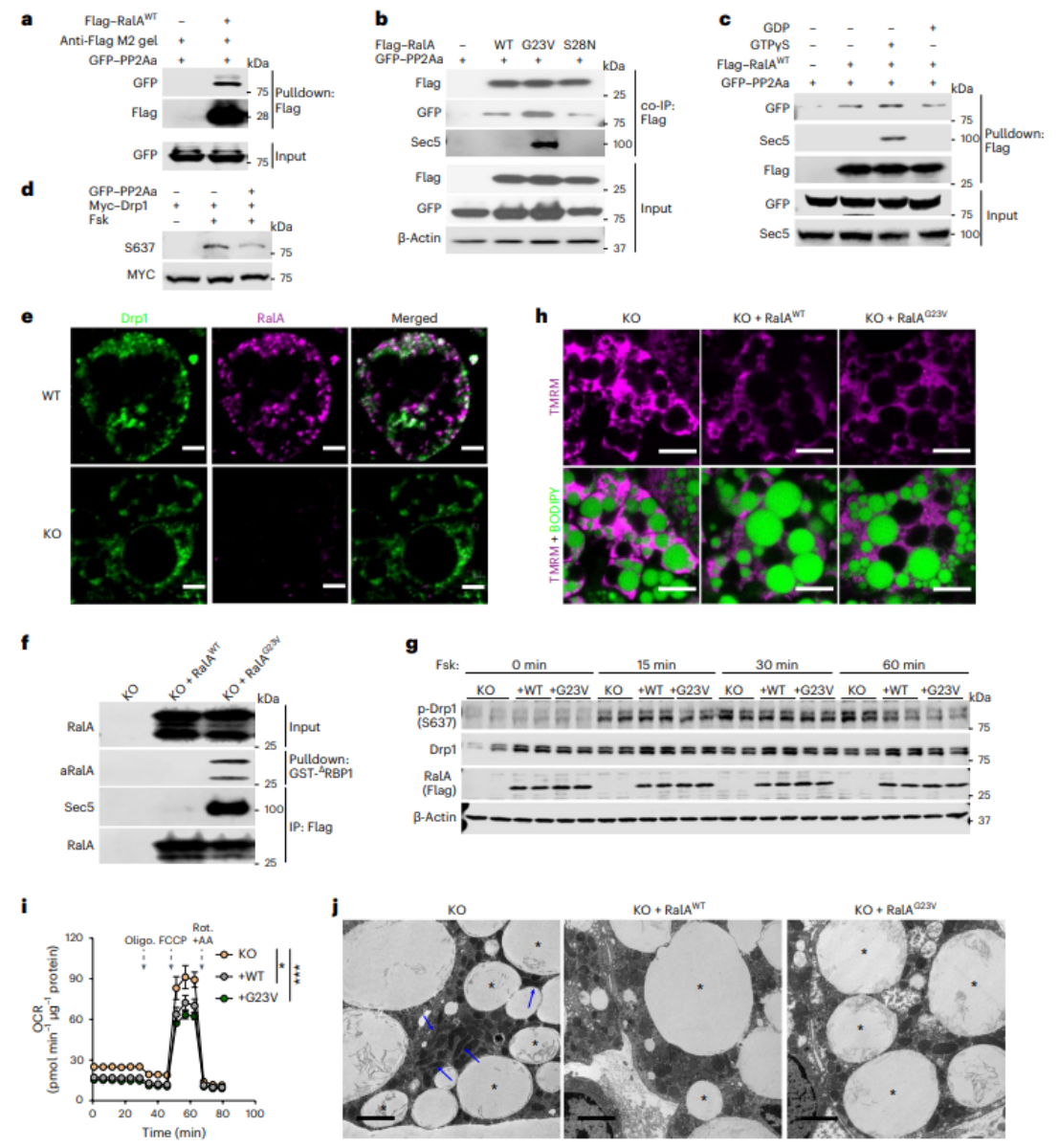
图6. RalA与Drp1和PP2Aa相互作用,促进Drp1 S637位点去磷酸化修饰
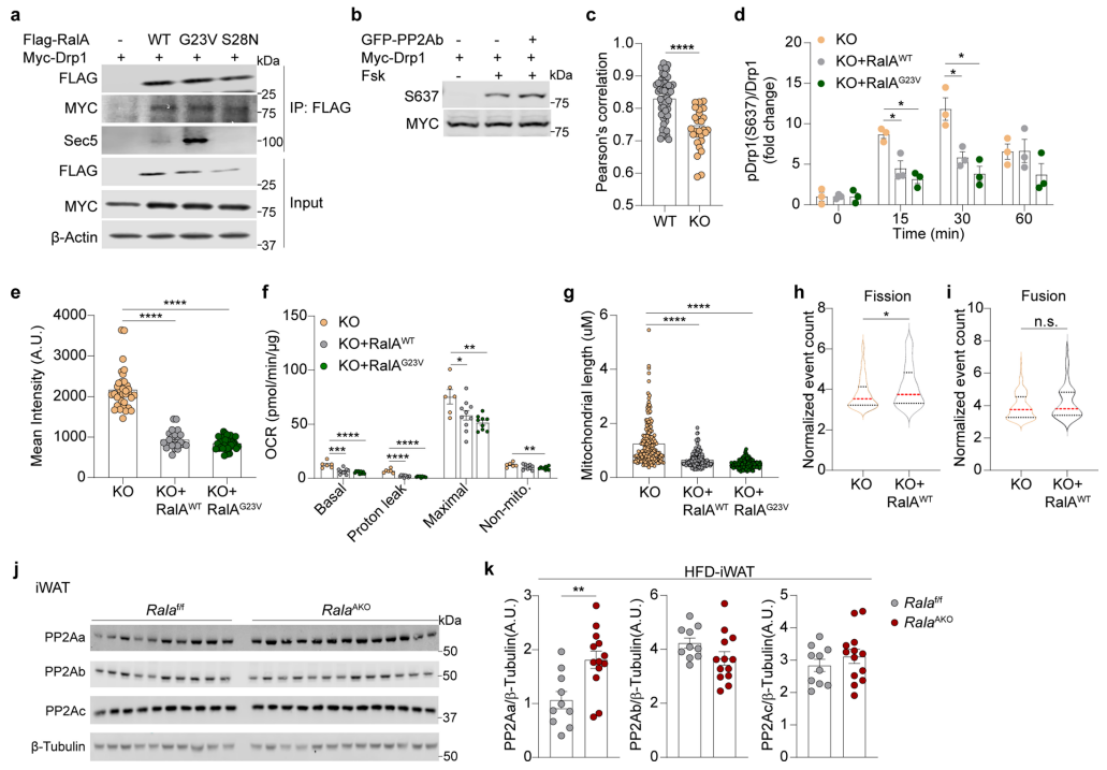
附图7. 敲除RalA可增加PP2Aa含量
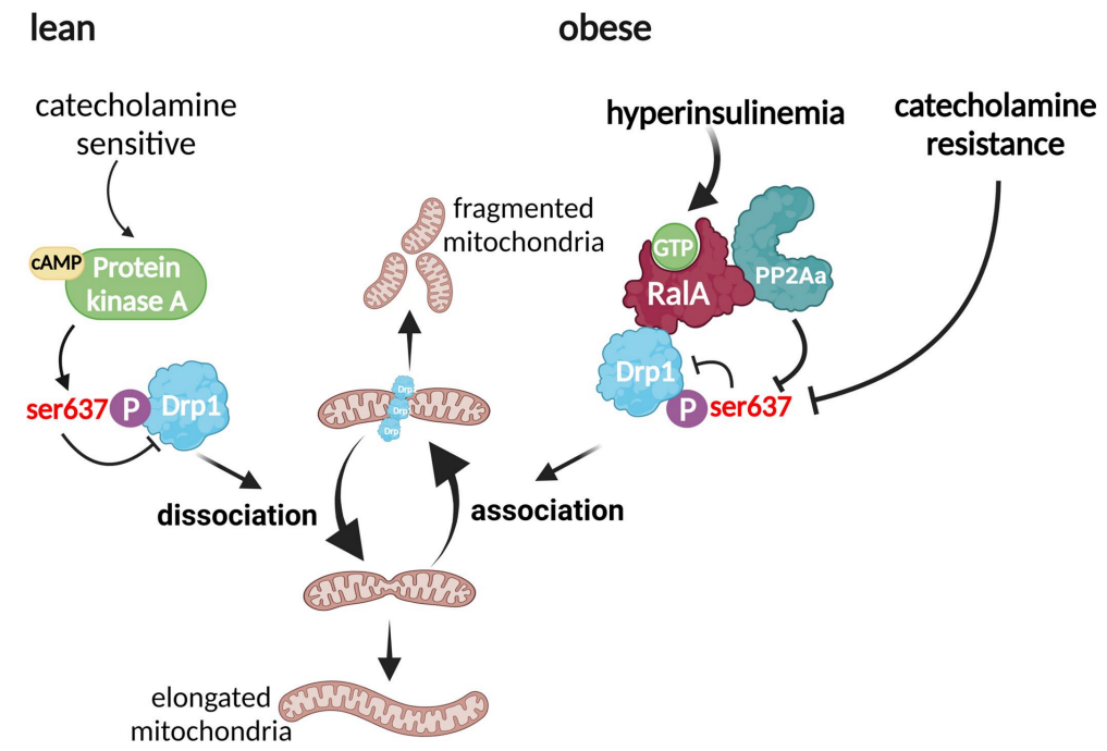
附图8. RalA调节肥胖脂肪细胞线粒体功能的机制模型
总结:
HFD小鼠白色脂肪细胞中RalA的表达和活性增加,致使线粒体断裂,进而导致氧化能力降低。靶向敲除白色脂肪细胞中的RalA可防止线粒体断裂,并通过增加脂肪酸氧化来改善HFD诱导的肥胖。从机制上讲,RalA通过促进Drp1 Ser637去磷酸化,提高Drp1活性,进而促进脂肪细胞线粒体断裂。综上所述,肥胖患者RalA持续升高会导致白色脂肪细胞线粒体功能障碍,进而致使体重增加和代谢功能障碍,对全身代谢产生深远影响。
链接:https://www.nature.com/articles/s42255-024-00978-0#Sec2
https://m.sciencenet.cn/blog-3483272-1427397.html
上一篇:好看的生化书——乘风破浪的戊糖磷酸
下一篇:代谢学人--Nature Metabolism:知否知否,肝EVs控糖高手