博文
Amber里建立力场prmtop和inpcrd方法
||||
大概流程及思路,参考:
http://ambermd.org/tutorials/pengfei/index.htm
具体操作方法:
目标:生成优化或者跑动力学时所需的力场prmtop文件和坐标inpcrd文件(prmtop文件同时包含了分子的力场参数信息和拓扑信息,力场参数是指分子的键长键角二面角等信息,拓扑信息是指分子的键连信息)从PDB Bank下载下来所需pdb后,去掉不需要的信息,以自己的实验体系为例,保留蛋白(一般选chain A)残基,配体,结晶水三部分。此时pdb结构文件中只有这三部分的重原子。对于后期跑动力学而言,需要所做的工作:加氢,加溶剂(抗衡离子和水),产生跑动力学所需prmtop,inpcrd文件。此处所讲即这几项工作如何进行。
生物体系,一般蛋白残基部分,结晶水和溶剂水,这三部分所需的力场采用amber自带的力场,如leaprc.ff03ua,leaprc.ff99SB等力场(amber软件在$AMBERHOME/dat/leap/cmd/路径下有很多版本力场,网上有介绍各版本力场的信息,根据需要挑选一个合适的力场版本。在此类力场中同时包含了水的力场,所以体系里的水分子不需要额外的leaprc力场文件,当然也有专门的水分子力场,根据需要自由选择)。有机小分子的力场一般采用Amber里的GAFF力场,即leaprc.gaff文件。
——————————————————-------------------------------
大概分如下几项:处理结构文件;生成配体小分子的模板;生成整体prmtop和inpcrd文件
第一项:处理结构文件
如前所述,做好蛋白,配体,独立的pdb文件,此时只包含重原子信息。
第二项:生成配体小分子模板
Amber中的antechamber程序是用来生成小分子模板的。
为什么配体小分子模板需要自己生成?
蛋白质中各氨基酸残基的力参数是预先存在的,但是很多有机小分子有很高的多样性,他们的力参数和静电信息不可能预存在库文件中,需要自己计算(如电荷)生成模板。
此处以总电荷为-1的配体分子为例:
关于配体,此处大概需要做的工作是:
1.计算原子电荷:
配体分子的原子电荷,可以选择bcc电荷,也可以选择resp电荷。不要求太高精确性,就采用bcc电荷,计算方法也简单。要求比较高的话,采用resp电荷
2.生成配体分子的mol2文件,frcmod文件。
这两个文件后续做力场时要用到。
上述2个工作具体操作起来如下:
0.首先准备好配体分子结构文件
准备好配体分子结构,以取名Lig.pdb为例,此时Lig.pdb只包含配体分子的重原子信息,需要进行加H工作,此工作采用amber里的reduce程序:
$reduceLig.pdb > Lig_addH.pdb
到此, Lig_addH.pdb已经加上H原子,这里需要check一下。用可视化软件如gsview查看加H后的pdb结构,check是否与自己所需的配体结构保持一致,不一致的地方在gsview里手动调整H原子的位置。假设需要调整,调整后的pdb结构命名:Lig_addH_Adjusted.pdb,之后用此结构去做高斯resp电荷,mol2文件等。
关于调整:需注意调整前后atom name的选取以及原子顺序,用reduce加H生成的pdb文件里包含一些不必要的信息,须删除,可手动,可用pdb4amber 程序进行。
1.计算配体分子的电荷
提前加载好antechamber路径。
(1)采用bcc方法拟合电荷:
$antechamber-i Lig_addH_Adjusted.pdb -fi pdb -o Lig.mol2 -fo mol2 -c bcc -nc -1 -pf y
解释:
-i 指定输入文件
-fi 指定输入文件类型
-o 指定输出文件
-fo 指定输出文件类型
-nc 指定体系所带总电量,此处为-1(如果没有-nc这一命令,则默认体系呈中性)
-pf y 不产生中间文件 (此命令可以不加,只是这样会产生一系列中间文件)
-c bcc 指定拟合原子电荷的方法,此处为采用bcc方法
生成的bcc电荷存储在Lig.mol2文件中:
(2)采用resp方法拟合电荷,方法:
http://blog.sciencenet.cn/home.php?mod=space&uid=3366368&do=blog&id=1079996
2.生成配体分子的mol2文件
如果采用bcc电荷,则上述bcc一步生成的mol2文件就可以直接用。但是,如果采用resp电荷,如下:
$antechamber-i Lig_addH_Adjusted.pdb -fi pdb -o Lig.mol2 -fo mol2 -nc -1 -pf y
此时,生成mol2文件:
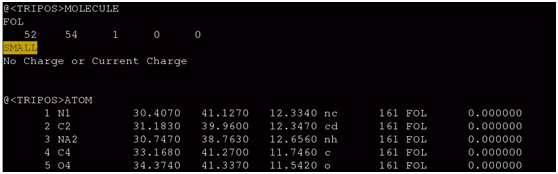
但是此处的mol2文件需要做以下修改:
将SMALL关键词的下一行替换成resp,图中最后一列0.000000替换成高斯计算出的resp电荷。到此,采用resp电荷的mol2文件准备好。
注:高斯计算resp电荷时原子顺序须与生成mol2文件所用的pdb文件中原子顺序保持一致。
3.生成配体分子的frcmod文件
采用amber里的parmchk程序:
$parmchk-i Lig.mol2 -f mol2 -o Lig.frcmod
到此,frcmod文件准备好。
第三项:生成模拟所需prmtop和inpcrd文件
所需文件:整体结构文件和上述生成的mol2文件和frcmod文件。
关于整体结构文件,一般是pdb文件(蛋白+配体,要不要结晶水自由决定),此处的结构文件只包含重原子信息,此处以all.pdb为例。
这里需注意:reduce自动加H后需要check,如果某些H原子所加位置需要调整,这一调整操作会引起重原子顺序发生变化,这里需要注意,要做到在整体结构文件all.pdb中的配体结构这一部分(未加H),配体重原子顺序应该与上述第0步中生成的Lig_addH_Adjusted.pdb结构中重原子顺序保持一致,一个简单的办法是删除Lig_addH_Adjusted.pdb中H原子信息,把剩余重原子信息替代all.pdb中配体部分。
为什么?
因为Lig_addH_Adjusted.pdb是手动调整过的(如果未调整,请忽略此处),比如重原子顺序以及atom name有可能与原始pdb bank下载下来的信息已不一致,但是所用的frcmod和mol2文件是根据Lig_addH_Adjusted.pdb生成的,所以此处应该保持一致。
生成prmtop和inpcrd采用amber软件包里的tleap或者xleap程序,两个程序功能相同,tleap是文字窗口,xleap是图形界面。
具体操作方法:
$tleap
到此,启动leap程序
>sourceleaprc.ff03ua
>sourceleaprc.gaff
到此,蛋白库文件leaprc.ff03ua和配体库文件leaprc.gaff加载进来了。
下面一步是调入配体分子的模板,补全库文件中缺失的参数:
>loadamberparamsLig.frcmod
到此,配体缺失参数补全。
下面一步是调入配体mol2文件:
>LIG= loadmol2 Lig.mol2
到此,mol2文件导入完毕。
需注意:LIG是Lig.mol2文件中配体分子的segname,即此处的变量名需与mol2文件以及整体结构all.pdb中配体分子的segname保持一致。
下面一步是调入整体结构:
>all= loadpdb all.pdb
注:如果结构是正确的,则导入后,屏幕出现如下信息:
total atoms in file: 1547
Leap added 1527 missing atoms according toresidue templates:
1507 H / lone pairs
20 unknown element
如果输入这个命令之后,屏幕上出现了大量的创建unit或者atom的信息,如下所示:
Createda new atom named: S33 within residue: .R
Createda new atom named: O35 within residue: .R
Createda new atom named: N34 within residue: .R
这说明上面一步的pdb文件处理没有做好,还需要重新处理,通常这种情况都发生在配体分子上面,有时则是因为蛋白质中存在特殊残基。
到此,结构导入进行,且已经加上H原子,如果没有其他所需操作,此时可以加离子和溶剂水了,为避免模拟中形成空穴,先加离子再加溶剂。
加离子,使体系保持中性:
>addionsall Na+ 0
addions加离子命令
Na+0 表示用Na+离子使体系保持中性(因为此时整个体系带负电)
若需要加阴离子,用Cl-。
(此处语法知识点:上述命令行里0表示中性,并不是加离子个数,但当此处为非0正整数时,含义则为所加抗衡离子个数。addions命令只能加整数个数的抗衡离子,如果体系所带电荷数为17.999,则用addions all Na+ 0这一命令只能加17个抗衡离子。显然如果为保持体系中性的话,这种操作的结果是不对的。这种情况应该具体指定所加离子个数,如对于17.999是需要加18个抗衡离子的,则用命令:addions all Na+ 18。总结:0表示最后结果呈中性,但此时要注意体系是否带整数个电荷量。非0值则表示所加离子具体个数,数值可任意指定,体系最后所带电量取决于此非0值和在加抗衡离子之前体系所带的电量)。
到此离子添加完毕,体系成中性。
加溶剂水分子:
>solvateboxall TIP3PBOX 10.0
solvatebox表示加上一个方形的周期水箱
all刚刚导入的存储有结构信息的变量
TIP3PBOX表示选择的水模板名称
10.0表示盒子大小,水箱半径
到此溶剂水添加完毕。
若无其他修饰,此时可以保存pdb,prmtop,inpcrd文件:
>saveamberparm all all_ionized.prmtop all_ionized.inpcrd
all_ionized.pdb 添加H,溶剂水,离子的结构信息,
all_ionized.prmtop 模拟所需的参数和拓扑文件
all_ionized.inpcrd 模拟所需的坐标文件
到此,力场建立完毕。
以上在tleap里的操作是一步一步来做,一个简单的办法是将以上所有操作写入leap.in文件,按如下操作一步完成:
$tleap-s -f leap.in > leap.out
leap.in内容:
sourceleaprc.ff03ua
sourceleaprc.gaff
loadamberparamsLig.frcmod
LIG= loadmol2 Lig.mol2
all= loadpdb all.pdb
addionsall Na+ 0
solvateboxall TIP3PBOX 10.0
savepdball all_ionized.pdb
saveamberparmall all_ionized.prmtop all_ionized.inpcrd
quit
到此,建立力场方法介绍完毕,分步做或采用leap.in来做,两种办法均可。all_ionized.prmtop,all_ionized.inpcrd是用于模拟所需的力场和坐标文件。此篇工作目的就是要拿到该两个文件。注意检查leap.log文件里有没有warning和error的信息,注意排查,确保所键力场信息正确。
在正式跑动力学之前可以check一下prmtop中拓扑参数和力场参数是否正确。方法:
查看拓扑信息:
在vmd里导入prmtop和结构文件(与prmtop配套的pdb文件),查看分子键连信息是否正确。
查看力场参数信息:
用prmtop简单跑一段动力学轨迹,查看键长键角等信息是否正确。
注:vmd导入prmtop文件时,vmd只提取里面的拓扑信息,力场参数如键角这些信息并不体现。
完!
https://m.sciencenet.cn/blog-3366368-1081381.html
上一篇:Amber里关于ambmask总结
下一篇:Amber里对轨迹文件mdcrd进行reimage处理