博文
代谢学人Cell Metabolism:肥胖和肿瘤的幕后黑手:SNORD46
||
代谢学人
Cell Metabolism:肥胖和肿瘤的幕后黑手:SNORD46
撰文 | 郑宇含 张婷 张彦康 李雨
编辑 | 孟美瑶
校对 | 刘梓棋


在过去几十年里,全球肥胖患病率急剧上升。肥胖一直被认为是许多慢性疾病的主要危险因素之一,包括2型糖尿病、心脏病和癌症。此外,有研究报道饮食诱导的肥胖可通过损害肿瘤微环境中CD8+T细胞功能,进而加速肿瘤的生长,这表明肥胖进一步阻碍了癌症患者基于免疫检查点的治疗效果。
IL-15是CD8+T细胞、NK细胞和上皮内淋巴细胞的细胞因子,参与维持淋巴细胞数量和活性,从而调控免疫细胞稳态。研究表明在多种肿瘤小鼠模型中,IL-15可通过提高NK细胞数量以及提高NK细胞活性,发挥抗肿瘤作用;并且体内高表达IL-15的NK细胞也表现出了更高的细胞毒性和活性;目前IL-15激动剂也已在临床试验中与免疫检查点抑制剂如PD-L1抗体联合使用治疗癌症。此外IL-15也可通过促进脂肪分解代谢,发挥抵抗肥胖的作用。有研究表明,对ob/ob小鼠给予IL-15治疗,可显著降低小鼠的脂肪组织重量,并且不影响小鼠食欲。然而IL-15抗肥胖的潜在分子机制还不清楚。
SnoRNAs是一类进化保守的非编码小向导RNA,其中大多数snoRNA参与调控其他RNA底物的化学修饰,如核糖体RNA的2-O-甲基化和假尿嘧啶化;一些snoRNA还参与调控mRNA的选择性剪接和转录后修饰。越来越多研究发现snoRNA的异常表达与疾病的发展有关,例如在男性中,SNORD38和SNORD48被确定为十字韧带损伤后骨关节炎发生发展的潜在血清生物标志物。然而,snoRNAs在肥胖及其相关代谢性疾病中的功能,目前还不清楚。
因此,在本文中,研究人员分析了肥胖血清中snoRNA水平变化,发现在肥胖人群的血清中,SNORD46水平显著增加,且SNORD46水平与BMI呈正相关。进一步研究发现饱和脂肪酸以依赖于C/EBPβ磷酸化的方式促进SNORD46的表达。此外,SNORD46可通过G11位点与IL-15相互作用,通过调控IL-15介导的非经典信号级联反应,抑制脂肪分解和棕色化进程,从而促进小鼠肥胖。最后,研究人员还发现SNORD46可抑制NK细胞中IL-15依赖性的自噬反应,导致肥胖条件下NK细胞抗肿瘤免疫功能受损。利用SNORD46抑制剂SNORD46pi治疗肥胖小鼠,可显著提高肥胖小鼠的NK细胞活力,也可促进肥胖条件下肿瘤细胞对CAR-NK细胞的敏感性,从而有效抵抗肿瘤的发展。总之,这项研究结果证明了snoRNA在肥胖和肿瘤免疫治疗中发挥着重要作用。

敲黑板啦!
1.snoRNA的表达与肥胖和免疫抑制相关
2.SNORD46抑制IL-15的经典和非经典信号通路
3.SNORD46通过抑制脂肪细胞分解,促进肥胖
4.snoRNA抑制剂可改善肥胖相关的免疫抵抗现象
结果:
1.血清SNORD46与肥胖和免疫抑制性肿瘤微环境有关
为了研究肥胖和肿瘤中的snoRNA水平变化,研究人员首先利用snoRNA阵列确定肥胖和非肥胖人群血清中的snoRNA拷贝数,发现在肥胖人群血清中SNORD46拷贝数上调(图1A,S1A-S1C)。随后,研究人员在BMI 16-78的382名受试者血清中进一步验证了SNORD46拷贝数变化,发现与BMI<25的受试者相比,BMI 30-40的受试者血清中SNORD46拷贝数显著上升,在BMI>40的受试者血清中SNORD46拷贝数进一步升高,而U6拷贝数无差异(图1B),且在肥胖人群中血清SNORD46拷贝数与BMI呈正相关(图S1D)。接着,研究人员检测了人和小鼠的不同组织中SNORD46表达量,发现在人和小鼠的脂肪组织中SNORD46表达量最高(图S1E)。此外,Northern blot结果显示血清SNORD46水平随BMI的增加而升高;而脂肪组织中,SNORD46表达量主要在肥胖患者的网膜(OM)和皮下(SubQ)脂肪上调,而BAT中几乎不表达SNORD46(图1C-D,S1F)。与不运动(0分钟/周)的人群相比,定期运动(>420分钟/周)的人群血清中SNORD46拷贝数显著减少(图1E)。同样的,不论受试者是否肥胖,血清SNORD46拷贝数都与受试者运动时间呈负相关,而血清U6拷贝数则无变化(图1F,S1G)。
随后研究人员利用TCGA数据库分析了肿瘤中snoRNA水平变化,发现与邻近正常组织相比,肿瘤中SNORD46水平升高(图S1H);并且,在SNORD46水平高表达的结肠癌(COAD)、乳腺癌(BRCA)和其他肿瘤中,肿瘤驻留性NK细胞浸润显著降低,且免疫表型评分(IPS)下降(图1G-H);在血清SNORD46水平高的乳腺癌患者肿瘤组织中,肿瘤驻留性CD8+T细胞和NK细胞也显著减少(由NCAM1/CD56标记)(图1I,1J)。总之,这些结果表明血清SNORD46水平与肥胖和免疫抑制性微环境相关。

图1.SNORD46水平与BMI和免疫抵抗有关

图S1. SNORD46表达谱
2.饱和脂肪酸通过C/EBPβ诱导SNORD46表达
为了确定调节SNORD46表达的转录因子,研究人员通过PICh分析(游离染色质片段的蛋白质组学分析)(小编注:PICh又被称为反向ChIP实验,其原理是使用特异性核酸探针与染色质杂交,杂交染色质被捕获在磁珠上,杂交体被洗脱,最终利用质谱技术鉴定所富集的蛋白质(Jérôme Déjardin,et al.Cell.2009.)。该实验不需要用到转录因子库进行分析),确定了一组与SNORD46启动子区相关的转录因子(图2A),并在人脂肪细胞(HAd)中逐一敲除了这些转录因子(图S2A,S2B),发现敲除C/EBPβ可显著抑制SNORD46的表达(图2B)。接下来,研究人员构建了C/EBPβ T235A突变体(模拟C/EBPβ去磷酸化修饰)和T235D/T235E(模拟C/EBPβ磷酸化修饰),发现C/EBPβT235A可显著降低人脂肪细胞上清液中SNORD46水平,而C/EBPβ T235D或T235E可显著上调SNORD46的表达水平(图2C,S2C);ChIP实验结果进一步表明C/EBPβ与SNORD46启动子区域的结合依赖于C/EBPβ磷酸化修饰(图2D);此外,在Cebpb KO小鼠脂肪细胞(MAds)中SNORD46表达显著降低,而过表达C/EBPβT188E突变体(模拟C/EBPβ磷酸化修饰)后可显著上调SNORD46的表达(图S2D-S2F),这表明C/EBPβ调控SNORD46的表达依赖于C/EBPβ磷酸化修饰。随后,研究人员对HFD诱导的肥胖小鼠进行跑步机训练(TE),发现与正常饮食小鼠相比,HFD饮食诱导的肥胖小鼠脂肪组织中C/EBPβ T235磷酸化水平显著升高,血清中SNORD46水平也显著上调,而TE后显著抑制了脂肪组织C/EBPβ T235磷酸化水平以及血清中SNORD46水平(图2E-G),研究人员推测脂肪酸可能参与调控了C/EBPβ磷酸化水平。随后研究人员用不同类型的饱和脂肪酸和不饱和脂肪酸处理人脂肪细胞,发现饱和脂肪酸可显著诱导人脂肪细胞中C/EBPβ T235磷酸化修饰,并上调细胞上清中SNORD46水平(图2H-I)。总之,这些结果表明饱和脂肪酸可通过促进C/EBPβ磷酸化,进而促进C/EBPβ对SNORD46的转录表达调控。

图2. C/EBPβ调节SNORD46的转录表达

图S2. C/EBPβ调节SNORD46的表达
3.SNORD46与IL-15相互作用并调节IL-15依赖性信号转导
接下来研究人员探究了SNORD46的调控机制。LC-MS分析结果显示SNORD46可与IL-15发生相互作用(图3A),随后研究人员对人和小鼠血清进行CLIP(交联免疫沉淀)分析,结果进一步证明了SNORD46可与IL-15相互结合,并鉴定出了SNORD46上IL-15的结合序列(图3B-C)。为了确定SNORD46中负责与IL-15结合的关键单核苷酸,研究人员构建了一系列SNORD46突变体,并回补到缺乏SNORD46基因的脂肪细胞中,发现G11C和G11U突变显著抑制了SNORD46与IL-15的相互结合,而G11A突变显著增强了SNORD46与IL-15的相互作用(图3D; S3A-S3D)。
为了确定IL-15中参与调控与SNORD46相互结合的关键氨基酸序列,研究人员利用Lip-MS(有限蛋白水解质谱技术)分析,发现在SNORD46 RNA单核苷酸存在下,IL-15 N端肽回收增加,表明IL-15的1-8氨基酸可能参与结合SNORD46(图3E)。随后为了明确调控IL-15与SNORD46结合的单氨基酸,研究人员构建了一系列IL-15 蛋白突变体,IL-15的D8A突变抑制了与SNORD46的结合,而D8S突变增强了与SNORD46的结合亲和力(图3F)。
为了进一步明确SNORD46与IL-15的结合机制,研究人员用IL-15的晶体结构(PDB:2Z3Q)构建了RNA蛋白对接的计算模型,发现SNORD46中G11的N1能与IL-15的N65形成氢键,而N65直接与IL-15的D8结合,从而形成稳定的SNORD46-IL-15复合物(图3G)。有趣的是,D8-N65的相互作用是两个α螺旋之间仅有的2个极性相互作用之一,表明D8-N65的相互作用对于维持IL-15与SNORD46的结合构象至关重要(图3G)。将SNORD46的G11突变为A后,羰基被胺基取代,胺基可以与N65形成额外的氢键(图3H),从而增加了SNORD45和IL-15之间的结合亲和力。将IL-15的D8位点突变为S后,羧酸基团被羟基取代(图S3E)。因为羧酸只能接受氢,而羟基可以做氢键的供体或受体,所以羟基可以与SNORD46的骨架形成氢键,同时维持与N65的相互作用(图S3E),从而增强SNORD46和IL-15的相互作用。研究人员在IL-15缺陷或SNORD46缺陷的人脂肪细胞分别回补野生型/突变体IL-15或野生型/突变体SNORD46,并进行RIP实验,验证了上述计算模型(图3I,3J,S3F-S3I)。
有研究表明,IL-15可以降低肥胖小鼠WAT组织重量,但其分子机制仍不明确。研究人员分别检测了对照或IL-15处理的人脂肪细胞(BMI<30)中的基因表达变化(GEO:GSE210203),结果显示IL-15处理健康受试者的脂肪细胞,显著下调了与脂肪形成和脂肪酸代谢相关的基因表达(图S3J)。总之,这些结果提示肥胖患者血清中SNORD46可能作为IL-15抑制剂,与IL-15相互作用并抑制IL-15的抗肥胖作用。

 图S3.SNORD46与IL-15相互作用
图S3.SNORD46与IL-15相互作用4.SNORD46 G11A突变促进肥胖
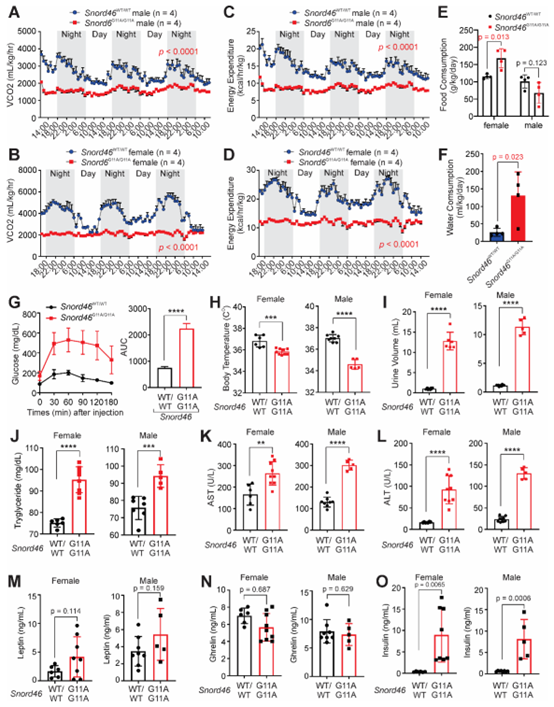
由于SNORD46在人类和小鼠之间高度保守,具有92.3%的同源性,研究人员用CRISPR-Cas9技术构建了含有小鼠Snord46基因单核苷酸变体(G11→A)的KI小鼠模型(图4A)。与WT和Snord46 G11A杂合突变小鼠相比,具有Snord46 G11A纯和突变的小鼠(Snord46G11A/G11A)体重、体脂和脂肪组织重量显著增加(图4B-4H)。组织学染色分析显示Snord46G11A/G11A小鼠脂肪组织脂滴变大,胰岛面积增大,肝脏脂质积累增加(图4I,4J)。此外,Snord46G11A/G11A小鼠耗氧量(VO2)和二氧化碳产生量(VCO2)减少,能量消耗降低(图4K,4L,S4A-S4D)。Snord46G11A/G11A雌鼠摄食量显著增加,而Snord46G11A/G11A雄鼠摄食量无差异,但Snord46G11A/G11A雄鼠水分摄入明显增加(图S4EF)。此外,Snord46G11A/G11A小鼠葡萄糖耐受下降,并表现出冷不耐受现象,且尿量、血清甘油三脂、AST、ALT均显著升高(图4M,S4G-S4L)。Snord46G11A/G11A小鼠的血清瘦素和生长激素释放肽水平与WT小鼠相似,但血清胰岛素水平显著升高(图S4M-S4O)。总之,这些结果表明SNORD46具有促肥胖作用,并且这与邻近基因RPS8的邻接性无关(图S5A-S5D)。

5.IL-15在脂肪细胞中介导非经典信号级联反应
为了探究Snord46G11A/G11A小鼠组织中IL-15介导的信号转导途径,研究人员鉴定了IL-15的结合蛋白,发现在WT小鼠的脑、心脏、肝脏、骨骼肌(SKM)、脾和肠中,IL-15与IL-15Ra和酪氨酸蛋白激酶JAK3结合,这是IL-15经典信号转导途径;而在WAT中,IL-15与酪氨酸蛋白激酶FER、单甘酯脂肪酶(MGLL)和CD36结合,触发潜在的非经典信号转导途径;而Snord46 G11A突变显著抑制了IL5的经典与非经典信号转到途径(图5A)。MS分析显示FER的Y402位点发生磷酸化,MGLL Y268位点发生磷酸化,CD36 Y370位点发生磷酸化(图S5E-S5G),因此研究人员设计了靶向人p-MGLL Y268和p-CD36 Y370的特异性磷酸化抗体(图S5H,S5I)。研究人员推测在配体结合时,IL-15受体复合物招募FER,从而介导下游信号级联反应,接下来研究人员利用IL-15处理MAd,发现转染对照的MAd中IL-15与IL-2Rβ、MGLL和CD36的结合,而在转染Snord46 G11A突变体的MAd中这种结合受到抑制(图5B)。
为了探究SNORD46突变体能否调控IL-15与IL-2Rβ的相互作用,研究人员用His标记的IL-15进行体外pull-down实验,发现SNORD46显著抑制了IL-15与IL-2Rβ的结合,且SNORD46 G11A突变体进一步抑制了IL-15与IL-2Rβ的结合,而SNORD46 G11C或G11U突变体对IL-15与IL-2Rβ的结合影响较小(图5C)。总之,SNORD46可抑制IL-15与受体复合物结合,而SNORD46 G11A可进一步抑制IL-15与受体的结合作用。
接下来研究人员进一步探究脂肪细胞中IL-15触发的FER/MGLL/CD36相关非经典信号途径。研究人员发现IL-15可诱导脂肪细胞中FER(Y402)、MGLL(Y268)和CD36(Y370)磷酸化修饰,而Il2rb缺失或Snord46 G11A突变则抑制这一磷酸化现象,且IL-15不能诱导NK细胞中上述的磷酸化修饰(图5D-5E),此外,IL-15可上调WAT棕色化相关蛋白的表达,如UCP1、PRDM16、CIDE-A、PPARγ,而IL2Rb缺失或SNORD46 G11A突变则抑制这些蛋白的表达(图5F)。与Snord46WT/WT相比,Snord46G11A/G11A小鼠WAT中UCP1和PPARγ水平也显著降低(图S5J,S5K)。
由于Snord46G11A/G11A小鼠表现出肥胖表型,研究人员检测了Snord46G11A/G11A小鼠脂肪细胞内/外的NEFA水平和细胞内TG水平,结果显示,IL-15处理显著降低了细胞内TG水平和细胞外NEFA水平,提高细胞内NEFA水平,然而敲减Il2rb或Snord46 G11A突变显著升高了细胞内TG水平和细胞外NEFA水平,降低了细胞内NEFA水平,且IL-15处理不影响细胞内TG水平和细胞内/外NEFA水平(图5G-5I)。据报道细胞内NEFA可促进脂肪细胞的FA氧化和耗氧率。因此,研究人员检测了Il2rb KO小鼠和Snord46G11A/G11A小鼠MAd的耗氧率(OCR),发现在WT小鼠的MAd中,IL-15处理可促进细胞OCR;而Il2rb缺失或Snord46 G11A突变则会抑制OCR,且IL5处理也不影响Il2rb缺失或Snord46 G11A突变小鼠MAd的OCR(图5J)。总之,这些结果表明IL-15通过调控非经典信号级联反应促进脂肪分解,进而缓解肥胖,而这一现象可被SNORD46抑制。
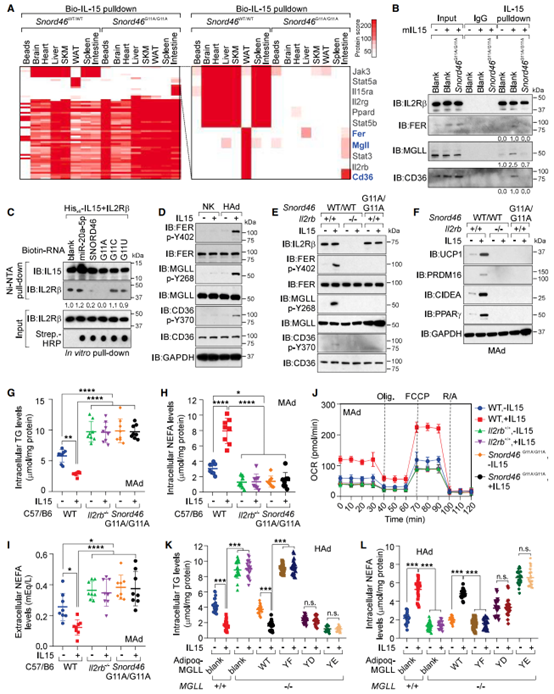
图5.IL-15引发脂肪细胞中由FER介导的信号级联反应,调节CD36和MGLL活性
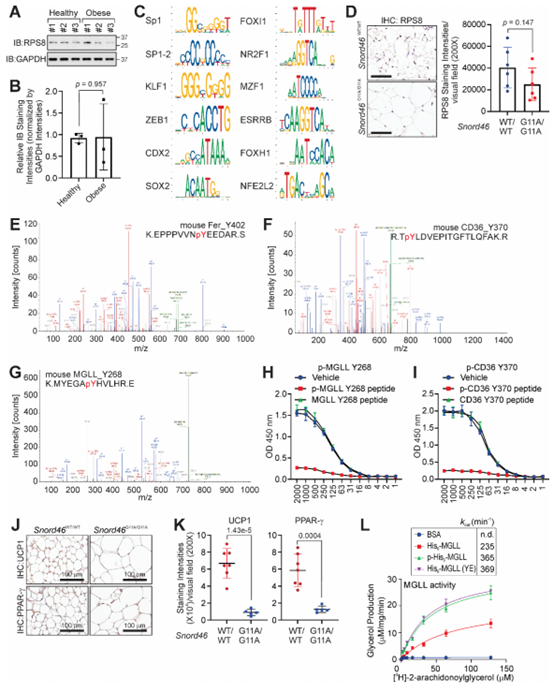
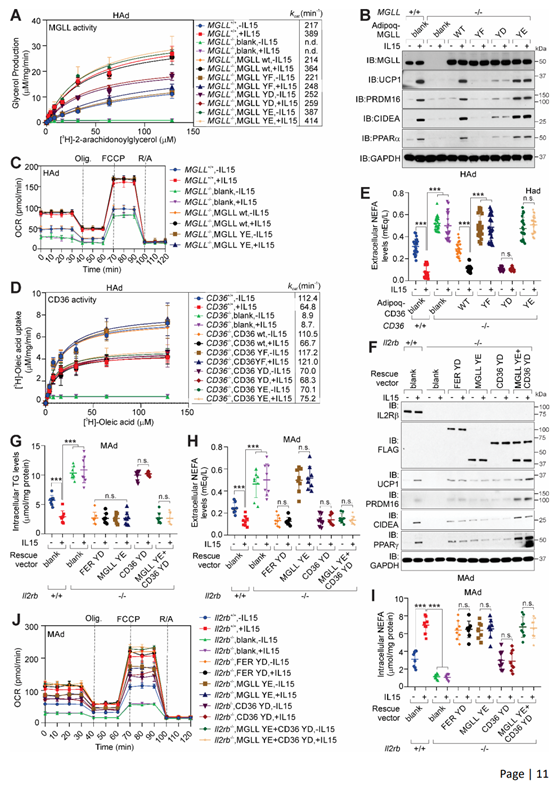
MGLL是脂肪分解过程的关键酶,可以将甘油单酯转化为甘油和FFA。研究人员发现与未磷酸化的MGLL相比,磷酸化的MGLL或MGLL Y268E(YE)突变体催化产生甘油的效率更高,表现出更高的酶活性(图S5L)。在人脂肪细胞中,研究人员发现IL-15可以促进甘油的生成,而MGLL缺失显著抑制了甘油的生成,回补外源性WT MGLL或MGLL YE突变体可恢复脂肪细胞中甘油的产生,但回补MGLL YD突变体仍显著抑制甘油的产生。(图S6A)。同样的,MGLL KO脂肪细胞内TG含量增加,NEFA减少,而回补外源性WT MGLL或MGLL YE突变体可以恢复IL-15诱导的NEFA产生(图5K,5L)。并且,MGLL KO脂肪细胞中UCP1、PRDM16、CIDE-A和PPARγ蛋白表达也被显著抑制,而回补WT MGLL或MGLL YE突变体后恢复了UCP1、PRDM16、CIDE-A和PPARγ蛋白的表达(图S6B)。此外,在MGLL KO脂肪细胞中回补WT MGLL或MGLL YE突变体也可恢复OCR水平(图S6C)。总之,这些结果表明IL-15可促使MGLL磷酸化修饰,从而调节脂肪分解过程。
CD36是一种脂肪酸转位酶,在与长链脂肪酸(LCFA)结合并将LCFA转运到细胞的过程中起重要作用。研究人员发现,IL-15处理或敲除CD36都会抑制脂肪细胞的FA摄取,在CD36 KO脂肪细胞中回补外源WT CD36或CD36 Y370F(YF)突变体可以恢复脂肪细胞摄取FA;而在CD36 KO脂肪细胞中回补CD36 Y370D(YD)突变体或CD36 Y370E(YE)可进一步抑制脂肪细胞摄取FA(图S6D)。此外,IL-15处理促使脂肪细胞胞外NEFA浓度降低,而CD36 KO脂肪细胞外NEFA水平升高,在CD36 KO脂肪细胞中回补外源WT CD36或CD36 YF突变体后可降低细胞外NEFA水平(图S6E)。总之,这些结果表明IL-15处理可促使CD36 Y370处磷酸化,从而抑制脂肪细胞摄取FA。
接下来,为了进一步明确SNORD46与IL-15依赖性FER/CD36/MGLL信号级联反应的联系,研究人员在Il2rb KO脂肪细胞中分别回补FER Y420D(YD)、MGLL YE、CD36 YD或MGLL YE+CD36YE突变体,发现Il2rb KO脂肪细胞中UCP1、PRDM16、CIDE-A和PPARγ蛋白水平显著下调,而回补FER Y420D可恢复UCP1、PRDM16、CIDE-A和PPARγ的蛋白水平,单独回补MGLL YE或CD36 YD突变体只能部分恢复UCP1、PRDM16、CIDE-A和PPARγ的蛋白水平,而同时表达MGLL YE+CD36 YD突变体时,可完全恢复UCP1、PRDM16、CIDE-A和PPARγ的蛋白水平(图S6F)。同样的,Il2rb KO脂肪细胞内TG水平和细胞外NEFA水平显著升高,细胞内NEFA水平显著降低;回补FER YD突变体可降低细胞内TG和细胞外NEFA水平,提高细胞内NEFA水平;单独回补CD36 YD突变体,对细胞内TG分解和NEFA的转运没有影响;而回补MGLL YE突变体或MGLL YE+CD36 YD突变体可显著降低细胞内TG和细胞外NEFA水平,提高细胞内NEFA水平(图S6G-S6I)。与此一致的是,在Il2rb KO脂肪细胞中回补MGLL YE突变体或MGLL YE+CD36 YD突变体也可以提高Il2rb KO脂肪细胞OCR(图S6J)。总之,这些结果表明IL-15在脂肪细胞中可触发FER/CD36/MGLL介导的非典型信号级联反应,从而调节脂肪分解和FA转运。
7
接下来,研究人员通过MS筛选了可体内抑制SNORD46与IL-15相互作用的SNORD46抑制剂,其中SNORD46 pi(SNORD46抑制剂#4)可特异性抑制SNORD46与IL-15的结合(图6A)。在肥胖人群的血清中,SNORD46 pi和IL-15的中和抗体(NAb)均可特异性抑制SNORD46与IL-15的相互作用(图6B,S7A-S7C)。此外,SNORD46 pi也显著抑制了IL-15与SNORD46 G11A突变体之间的相互作用(图6C)。
在没有IL-15处理的情况下,与BMI<30人群相比,BMI>40人群脂肪细胞中FER(Y420)、MGLL(Y268)和CD36(Y370)磷酸化水平降低(图6D,第9列与第1列对比);在IL-15处理的条件下,BMI<30人群的脂肪细胞中FER(Y420)、MGLL(Y268)和CD36(Y370)磷酸化水平显著升高(图6D,第2列与第1列对比),而BMI>40人群脂肪细胞中FER(Y420)、MGLL(Y268)和CD36(Y370)磷酸化水平并未发生变化(图6D,第10列与第9列对比)。SNORD46 pi处理BMI<30人群脂肪细胞,可以增强脂肪细胞对IL-15的敏感性,从而上调IL-15诱导的FER(Y420)、MGLL(Y268)和CD36(Y370)磷酸化水平,以及UCP1、PDRM16、CIDE-A和PPARα的蛋白质水平(图6D,第4列与第3列对比);此外,SNORD46 pi处理也可增强BMI>40人群脂肪细胞对IL-15的敏感性,从而上调IL-15诱导的FER(Y420)、MGLL(Y268)和CD36(Y370)磷酸化水平,以及UCP1、PDRM16、CIDE-A和PPARα的蛋白质水平(图6D,第12列与第11列对比)。
接下来,研究人员检测了不同BMI人群脂肪细胞内TG、NEFA和细胞外NEFA水平,结果表明,BMI<30时,IL-15诱导脂肪细胞内TG和细胞外NEFA降低,细胞内NEFA升高;BMI>40时,IL-15对细胞内TG、NEFA和细胞外NEFA水平无显著影响(图6E-6F,S7D);然而,SNORD46 pi处理显著提高了BMI>40脂肪细胞对IL-15的敏感性,进而降低脂肪细胞内TG和细胞外NEFA水平,提高细胞内NEFA水平(图6E-6F,S7D)。同样的,与BMI<30相比,BMI>40的脂肪细胞OCR水平显著降低,而SNORD46 pi处理可以显著提高BMI>40的脂肪细胞OCR(图S7E)。
此外,与Scramble序列、奥利司他和利拉鲁肽(2种FDA批准的抗肥胖药物)相比,Snord46 pi治疗可显著降低Snord46G11A/G11A小鼠体重,并改善葡萄糖稳态和肝脏脂肪变性现象,WAT中UCP1和PPARγ蛋白水平也显著提高(图6G-H,S7F-I)。总之,这些结果表明,SNORD46 pi可能通过抑制SNORD46-IL-15的相互作用,从而缓解肥胖。

图6. SNORD46抑制剂缓解肥胖

8
8. 靶向抑制SNORD46可恢复抗肿瘤免疫微环境
有研究表明,肥胖患者NK细胞的细胞毒性降低。因此,为了探究SNORD46水平的升高是否会降低肥胖患者NK细胞的细胞毒性,研究人员分别从BMI<25人群(其NK细胞称为正常NK)和BMI>35人群(其NK细胞称为肥胖NK)的外周血单核细胞(PBMC)中分离NK细胞,发现与正常人群相比,肥胖人群的NK细胞数量显著下降;同样,与WT小鼠相比,Snord46G11A/G11A小鼠外周血中的NK细胞也显著减少(图S8A-C)。对正常和肥胖NK细胞的转录谱分析表明,高表达SNORD46的肥胖NK细胞中炎症反应、TNFα信号和JAK/STAT3信号相关基因表达显著下调(图7A,S8D-S8G);并且肥胖NK细胞中自噬相关基因也显著下调(图7B),提示肥胖人群NK细胞的自噬反应受损;且与正常BMI人群相比,BMI>35人群NK细胞中LC3A/B(自噬标志蛋白)表达水平显著降低(图7C)。同样的,研究人员利用自噬诱导剂TBM处理人NK细胞或Snord46G11A/G11A小鼠的NK细胞,发现与正常NK细胞或WT小鼠NK细胞相比,肥胖NK细胞或Snord46G11A/G11A小鼠NK细胞活力均显著下降;而SNORD46 pi处理则显著提高了肥胖NK细胞活力和Snord46G11A/G11A小鼠NK细胞活力(图7D-7G)。
接下来研究人员进一步探究了SNORD46pi能否增强肥胖NK细胞的细胞毒性,从而提高肥胖状态下肿瘤对CAR-NK细胞治疗的敏感性。研究人员构建了2种CAR-iPSCs(诱导多能干细胞),分别表达EGFR-CAR和CD133-CAR,并分化为具有高特异性和高纯度的NK细胞(图S8H,S8I)。EGFR-CAR-iPS-NK细胞对人三阴性乳腺癌(TNBC)细胞(MDA-MB-231)表现出抗肿瘤免疫性,而CD133-CAR-iPS-NK细胞对人结直肠癌细胞(HT-29)表现出抗肿瘤免疫性(图S8J-S8L)。在正常喂养条件下,CD133-CAR-iPS-NK细胞治疗能显著抑制小鼠体内HT-29肿瘤的生长;但在HFD喂养条件下,CD133-CAR-iPS-NK细胞治疗对小鼠HT-29肿瘤的生长无明显影响;而SNORD46pi与CD133-CAR-iPS-NK细胞联合治疗,可显著提高HT-29细胞对CD133-CAR-iPS-NK细胞的敏感性,从而显著抑制HT-29肿瘤的生长(图7H)。同样的,在正常喂养下,EGFR-CAR-iPS-NK细胞治疗可显著抑制小鼠体内MDA-MB-231肿瘤的生长,但在HFD喂养下,EGFR-CAR-iPS-NK细胞治疗对MDA-MB-231肿瘤的生长无明显影响,而SNORD46pi治疗可显著促进MDA-MB-231细胞对EGFR-CAR-iPS-NK细胞的敏感性(图7I)。随后研究人员检测了正常和肥胖小鼠肿瘤中CAR-iPS-NK细胞水平,发现在HFD小鼠中,MDA-MB-231和HT-29肿瘤中NK细胞水平均显著下降,而SNORD46pi治疗可显著提高正常饮食和HFD小鼠肿瘤中NK细胞水平(图7J,7K)。综上,这些结果表明,基于snoRNA的治疗方法可精确靶向治疗肥胖和肥胖相关免疫疾病。

图7.SNORD46抑制剂提高肥胖下CAR-NK细胞的抗肿瘤免疫性

图S8. SNORD46抑制剂恢复肥胖NK细胞的自噬功能
总结
本文中,研究人员发现血清SNORD46水平与BMI和肿瘤微环境相关,并且血清SNORD46能抑制IL-15信号转导。机制上,研究人员发现饱和脂肪酸(SAFA)以依赖于C/EBPβ磷酸化的方式促进SNORD46的表达,SNORD46可通过G11与IL-15相互作用,而SNORD46与IL-15的结合会抑制IL-15介导的非经典信号级联反应,从而抑制脂肪分解和棕色化过程,促进小鼠肥胖。此外,研究人员还发现SNORD46能抑制NK细胞自噬反应,进而降低NK细胞的抗肿瘤免疫性。最后研究人员筛选出SNORD46抑制剂SNORD46pi,具有抵抗肥胖,提高肥胖小鼠NK细胞活力以及促进肿瘤对CAR-NK细胞的敏感性的治疗作用。总之,本项研究证明了snoRNA在肥胖调控中的重要性,并证明了snoRNA抑制剂用于肥胖和肥胖相关免疫抵抗治疗的潜能。

关注微信公众号代谢学人
了解更多代谢前沿资讯

https://m.sciencenet.cn/blog-3483272-1395451.html
上一篇:代谢学人Nature Metabolism:血管有难,棕色脂肪来也!
下一篇:代谢学人Nature Metabolism:产热脂肪与交感神经之“锌”的秘密