博文
代谢学人Nature :八角笼中——相分离抑制铁死亡新机制!
||
代谢学人
Nature:八角笼中——相分离抑制铁死亡新机制!
撰文 | 夏志蕊 朱丽君 刘爽 曹玉香
编辑 | 孟美瑶
校对 | 刘爽

背景介绍
铁死亡是一种以铁依赖性脂质过氧化为特征的,非凋亡性细胞死亡的代谢形式。由于铁死亡与人类疾病(如神经退行性疾病、冷暴露期间的组织损伤、缺血再灌注损伤和癌症)高度相关,因此引起了人们极大的兴趣。特别是在恶性肿瘤的背景下,触发铁死亡已成为一种非常有前途的方法,它与癌症免疫疗法具有协同作用,甚至可以杀死耐药和转移性肿瘤。最近的研究表明,FSP1(铁死亡抑制蛋白1)是铁死亡防御系统的备份系统(小编注:细胞中有多个对抗铁死亡的防御途径,其中最主要的一个是由谷胱甘肽过氧化物酶4(GPX4)所介导的,通过谷胱甘肽(GSH)特异性催化过氧化脂质来抑制铁死亡的发生。FSP1是一种不依赖谷胱甘肽的新型强效铁死亡抑制因子,通过将细胞膜上的泛醌转化为其还原型泛醇,泛醇能抑制过氧化反应并阻止铁死亡。),尽管靶向FSP1已被认为是癌症治疗的一个有吸引力的药物靶点,但一直缺乏体内有效的FSP1抑制剂。已有的FSP1特异性抑制剂iFSP1并不适合在体内使用,其在高浓度时会出现脱靶效应,在药物化学开发方面潜力有限,不适合作为抗癌药物进一步开发。因此,迫切需要新一代高效的FSP1抑制剂用于肿瘤治疗。
近期发表在Nature杂志上的一篇题为“Phase separation of FSP1 promotes ferroptosis”的文章中,研究人员通过小分子文库筛选到了一类3-苯基喹唑啉酮类化合物(icFSP1)作为FSP1的抑制剂,与现有的FSP1抑制剂不同,icFSP1不是通过竞争性抑制酶活性,而是通过促使FSP1从细胞膜中重新定位和形成FSP1凝聚体的方式诱导铁死亡,同时与GPX4的抑制协同作用。这些由icFSP1诱导的FSP1凝聚体表现出类似相分离的特性,这是一种调节生物活性的新兴机制。

敲黑板啦!
1.icFSP1(新一代FSP1抑制剂)与iFSP1促进铁死亡的机制不同;
2.icFSP1在铁死亡前触发FSP1的亚细胞重新定位和相分离;
3.icFSP1介导FSP1的相分离需要N-末端肉豆蔻酰化、氨基酸残基、无序区和低复杂性区;
4.icFSP1抑制肿瘤生长
研究结果
1.icFSP1引起铁死亡
为了寻找能在体内适用的新型FSP1抑制剂作为潜在的药物对抗难以治疗的癌症,研究人员验证了大约10000种小分子化合物,使用化学信息学工具来预测其物化性质和药物相似性。进一步通过苗头化合物验证(小编注:也称Hit验证,一种通过高通量筛选技术初步筛选得到具有一定生物活性化合物的研究方法)确认了一类3-苯基喹唑啉酮类化合物(以先导化合物icFSP1为代表)是一类强效的FSP1药理抑制剂(图1a)。初步的构效关系研究尚未发现比icFSP1更有效的化合物(辅图1a)。
随后发现icFSP1对4-羟基塔莫西芬(TAM)诱导的稳定高表达人FSP1(hFSP1)的Gpx4基因敲除小鼠Pfa1细胞和人纤维肉瘤HT-1080细胞系引起明显的脂质过氧化和相关的铁死亡特征(图1b-g和辅图1b)(小编注:TAM诱导的Gpx4基因敲除的小鼠永生化成纤维细胞构建是利用CRERT2/loxP系统通过TAM诱导的Cre重组酶的激活来实现基因组Gpx4的缺失。GPX4是限制性GSH依赖酶,缺乏GPX4的小鼠会在发育阶段死亡,研究中的条件性敲除小鼠利用MERCreMeR(MER是突变雌激素受体)转染细胞,MERCreMER通过与热休克蛋白90形成复合物而保留在细胞质中,只有在细胞培养基中加入TAM后,MERCreMER才会从抑制复合物中释放出来,并转运到细胞核中进行Cre介导的重组,在72h内使GPX4转录水平下降)(小编再注:雌激素受体(ER)是雌酮、雌二醇等女性荷尔蒙的细胞内蛋白受体,分为核雌激素受体和膜雌激素受体。在正常小鼠体内,没有雌激素的情况下,核雌激素受体跟HSP90结合在一起。由于HSP90的阻挡,雌激素受体无法进入细胞核,只能游荡于细胞质内。雌激素出现后,雌激素会跟核雌激素受体结合,并将HSP90从雌激素受体上排挤走。HSP90消失后,雌激素受体便可进入细胞核,调控位于核内的基因的表达。研究人员通过基因编辑,将雌激素受体的配体结合域和Cre酶融合在一起。此时,没有雌激素时,Cre酶与雌激素受体配体结合域和热休克蛋白绑定在一起,停留在细胞质内,Cre酶无法进入细胞核启动目标基因表达。当雌激素出现时,热休克蛋白被排挤掉,Cre酶和雌激素受体的结合体蛋白进入细胞核,Cre酶也就可以发挥作用。另外,研究人员为了排除内源雌激素的影响,使用了突变的雌激素受体。经过多重药物筛选后,发现突变后的雌激素受体不再结合体内的雌激素,只对TAM这种外源药物有亲和力。也就是说,只有当TAM出现时,Cre酶才能入核调控基因表达。同时,由于与Cre酶融合的只是雌激素受体的配体结合域,因此不会触发雌激素受体相应的基因调控。参考文献:[1] Kam MK, et al. PLoS One. 2012;7(5):e35799.)。icFSP诱导的细胞死亡可以通过铁死亡抑制剂来挽救,但不能通过靶向其他形式的细胞死亡的抑制剂来挽救,从而证实了其对铁死亡的特异性。对于靶向FSP1引起的细胞死亡(如癌细胞)通常需要和其他类型的铁死亡典型诱导剂共同作用(比如system Xc-抑制剂erastin、谷氨酸半胱氨酸连接酶(GCL)抑制剂I-丁硫氨酸亚砜胺(BSO)、GPX4抑制剂RSL3、ML210、FIN56以及铁氧化化合物FINO2),而与其他类型的诱导剂如细胞凋亡诱导剂staurosporine,细胞程序性坏死诱导剂TNFα+smac mimetic(S)+z-VAD-FMK(Z),细胞焦亡诱导剂nigericin等都不能引起细胞大量死亡(辅图1c-e)。因此,令人惊讶的是,与一组不同的人类癌细胞系相比(辅图1c),单独使用icFSP1或多西环素诱导的FSP1敲除HT-1080细胞72小时足以引发铁死亡(图1d和辅图1f-h) (小编注:多西环素(四环素衍生物)诱导的条件性基因敲除系统包含两个互补系统,tTA(四环素控制的反式激活蛋白)依赖和rtTA(反式四环素控制的反式激活蛋白)依赖的基因敲除系统,现在又称为Tet-Off系统和Tet-ON系统。在这两个系统中,四环素或其衍生物多西环素控制转录激活子tTA或rtTA与启动子Ptet结合,从而调控下游基因的表达。不同的是:Tet-Off系统:tTA与Ptet启动子结合不需要四环素或Dox,即可启动转录;但加入Dox后,形成Dox-tTA复合物,构象改变而不与启动子结合,阻碍目的基因表达。Tet-On系统:rtTA与启动子结合需要四环素或Dox;加入Dox后,形成Dox-rtTA复合物与启动子Ptet结合,才能启动目的基因表达。在本文中,为了获得Dox诱导的敲除细胞,研究人员使用Tet-off系统,通过设计靶向目的基因关键外显子的sgRNA,使用含有靶向FSP1的sgRNA的慢病毒感染HT–1080细胞,使用嘌呤霉素筛选并建立单细胞克隆)。为了研究icFSP1是否具有脱靶效应,我们将HT-1080和HEK293T细胞以及原代外周血单个核细胞(PBMCs)分别暴露于FSP1抑制剂(icFSP1和iFSP1) 72小时和24小时。与iFSP1相比,icFSP1即使在较高浓度下也没有表现出脱靶效应(辅图1h-j)。除此之外,当细胞与GPX4抑制剂共孵育时,用增加浓度的icFSP1处理FSP1敲除细胞没有显示出任何协同效应(辅图1k,l),这表明icFSP1应该是一种选择性FSP1抑制剂。
为了研究icFSP1是否也在非人类细胞中诱导死亡,我们在研究中纳入了两种不同的小鼠细胞系和一种大鼠成纤维细胞系(分别为4T1、B16F10和Rat1),结果表明,与RSL3或GPX4敲除共同处理未能协同杀死这些细胞(辅图2a-g)。与此一致,在稳定过表达小鼠FSP1的Pfa1细胞中,icFSP1不影响脂质过氧化和细胞活力(辅图2h,i),表明icFSP1特异性抑制了人类细胞。此外,我们在Pfa1细胞中测试了FSP1的其他同源物,过表达来自鸡和非洲爪蟾的FSP1。虽然FSP1过表达完全阻止RSL3诱导的铁死亡,但icFSP1仅在过表达hFSP1的细胞中降低细胞活力(辅图2j-l)。因此,这些数据强化了icFSP1是hFSP1特异性抑制剂的概念。
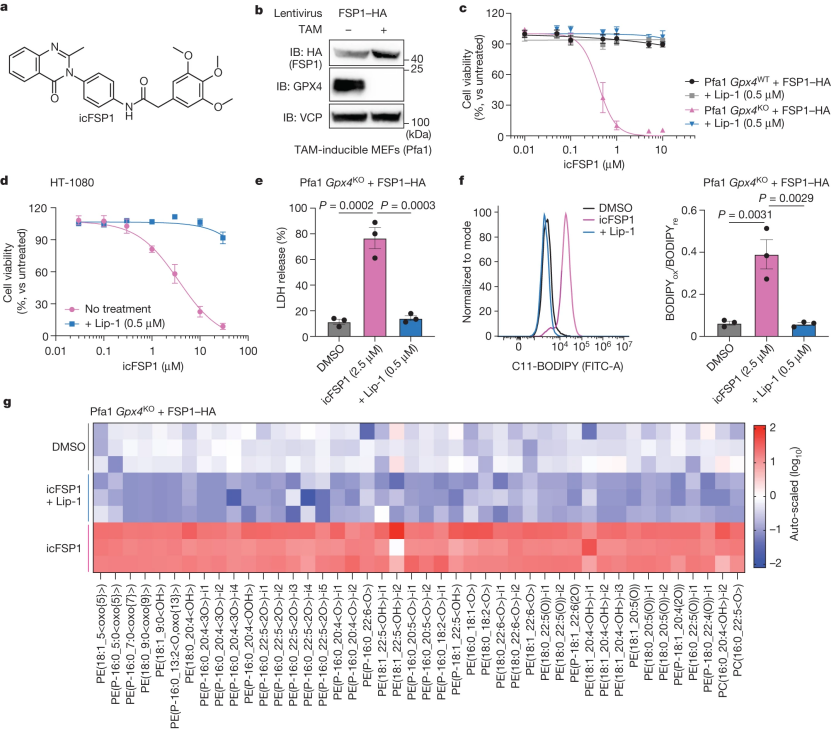
图1 icFSP1和GPX抑制剂协同抑制铁死亡
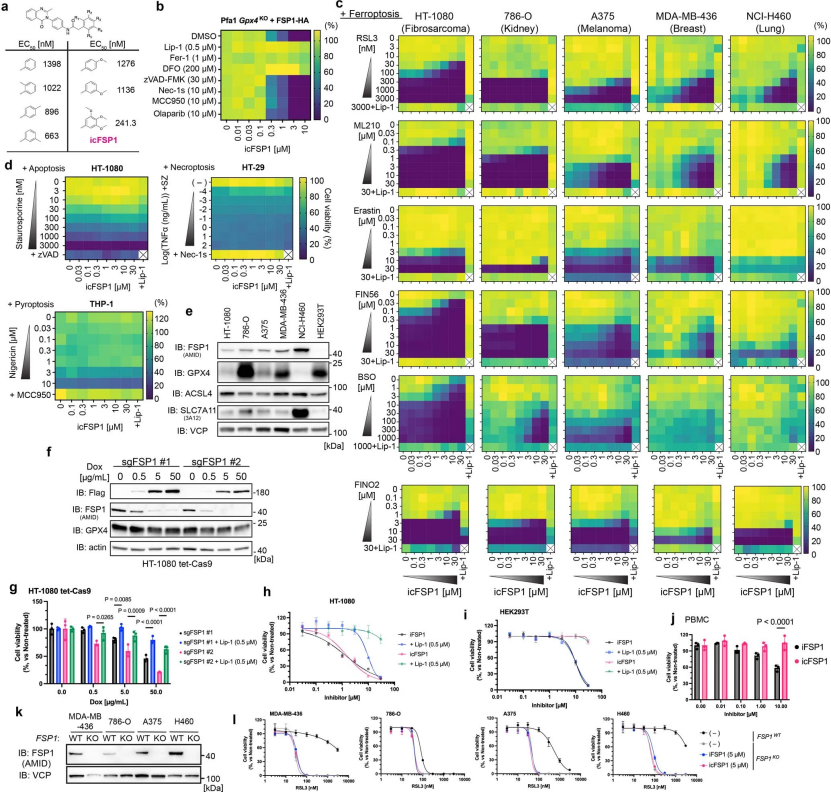
辅图1 在多种人癌细胞中,icFSP1与其他铁死亡诱导剂协同作用

辅图2 icFSP1不能抑制鼠源FSP1
2.icFSP1诱导FSP1凝聚
为了研究这些新一代FSP1抑制剂的作用机制,研究人员利用建立重组hFSP1酶的实验方法,在体外测量了FSP1的直接抑制活性(图2a)。据报道,在非细胞系统中iFSP1抑制了hFSP1的酶活性,而icFSP1在低微摩尔范围内并不抑制hFSP1的活性,但它明显影响了过表达hFSP1的Pfa1细胞的细胞活力和脂质过氧化能力(图2b和辅图3a-k)。事实上,体外实验中估计的icFSP1的IC50(半抑制浓度)比在Pfa1细胞中观察到的EC50(半最大有效浓度)高出100倍以上(图2b和辅图3i)。这些结果有力地证明,与iFSP1相比icFSP1抑制细胞中hFSP1的活性机制不同(小编注:IC50指被测量的拮抗剂的半抑制浓度,指示某一药物或者物质(抑制剂)在抑制某些生物程序(或者是包含在此程序中的某些物质,比如酶,细胞受体或是微生物)的半量。研究人员通过检测hFSP1活性的变化(图2b),发现相同浓度的iFSP1和icFSP1对hFSP1的活力影响不同,即IC50不同,说明二者对hFSP1的作用机制不同)。为了阐明其作用机制,研究人员首先分析了icFSP1是否会降低hFSP1的表达水平。利用icFSP1分别处理过表达hFSP1的H460或HT-1080细胞48小时和72小时后免疫印迹结果显示,icFSP1处理不影响hFSP1的表达水平 (辅图3l–n)。接下来,研究人员认为icFSP1可能通过将其从脂质膜上分离而改变了hFSP1的亚细胞定位,从而阻止了其通过泛醌或维生素E(或K)清除磷脂自由基的抗铁蛋白功能。为此,研究人员使用融合了增强型绿色荧光蛋白和链霉亲和素的hFSP1(hFSP1-EGFP-Strep)(小编注:Strep是质粒中常用的多肽标记物,与His-Tag和FLAG-Tag相似,作为一个标记物存在。Strep-tag亲和标签系统的纯化条件比较宽泛,不但在普通缓冲液(例如磷酸盐缓冲溶液,PBS)下可以和Strep-Tactin层析介质结合,而且使用2.5mM的脱硫生物素就可快速的将融合蛋白洗脱下来。研究人员使用此标签标记hFSP1,也是为了后面纯化hFSP1做准备)。在Pfa1细胞中稳定过表达,并经过icFSP1处理后监测其定位。与iFSP1不同,icFSP1显著改变了hFSP1-EGFP-Strep的亚细胞定位,这可以通过明显的颗粒和细胞凝聚物的出现来说明(图2c和辅图3o)。这些凝聚物在细胞中以时间依赖的方式积累(图2d),并且只出现在表达hFSP1的细胞中,而不出现在表达mFSP1的细胞中(辅图3p),证实了icFSP1对人类同源物具有特异性。为了检验hFSP1亚细胞定位的改变是否会诱导铁死亡,研究人员建立了稳定过表达hFSP1与蓝色荧光蛋白(BFP) 融合的GPX4敲除的Pfa1细胞。hFSP1-BFP信号通过Liperfluo(一种脂质过氧化氢传感器)和碘化丙啶共同染色的活细胞成像来监测,碘化丙啶只有在质膜发生渗漏时才能将细胞核染色。在用icFSP1处理表达hFSP1–BFP的细胞后,诱导产生凝聚物然后发生Liperfluo氧化,细胞中的脂质过氧化信号逐渐增加,直到细胞碘化丙啶呈阳性(细胞膜破裂的指标)(图2e)。这些结果表明,FSP1亚细胞定位的改变先于脂质过氧化和铁死亡。
拓展阅读
维生素K通过FSP1抵抗铁死亡
维生素K是一种必需的微量营养素,参与多种生理过程,作为肝脏维生素K依赖性凝血因子(包括因子II、VII、IX和X) γ-羧化的辅助因子,其对凝血级联反应至关重要。此外,与泛醌类似,维生素K是具有氧化还原活性的萘醌,在植物和细菌中作为ATP生成的电子载体,最近被证明其可以防止铁死亡。维生素K对苯二酚(VKH2)在体内具有促进凝血的功能,通常情况下,维生素K环氧还原酶(VKOR)将维生素K还原为VKH2,从而维持VKH2 的水平。VKH2可作为一种强亲脂性抗氧化剂,并通过在脂质双分子层中捕获氧自由基来防止铁死亡。
FSP1可以通过消耗NAD(P)H,将线粒体外泛醌(CoQ10)还原为泛醇。泛醇反过来作为一种有效的亲脂性自由基捕获抗氧化剂,直接减少膜中的脂质自由基,从而防止脂质过氧化反应。因此,FSP1可以以一种类似于泛醌的方式——作为维生素K还原酶减少维生素K,从而再生VKH2,避免非典型维生素K循环中的脂质过氧化和相关的铁死亡。并且,MK-4对苯二酚在体外抑制脂质过氧化的作用比还原形式的泛醌更有效,在这种情况下,补充维生素K可能是减少神经退行性疾病和其他疾病症状的一种方法。

铁死亡的Gpx4途径和Vitamin K途径
参考文献
[1] Mishima E, et al. Nat Metab. 2023 Jun;5(6):924-932.
[2]Mishima E, et al. Nature. 2022 Aug;608(7924):778-783.
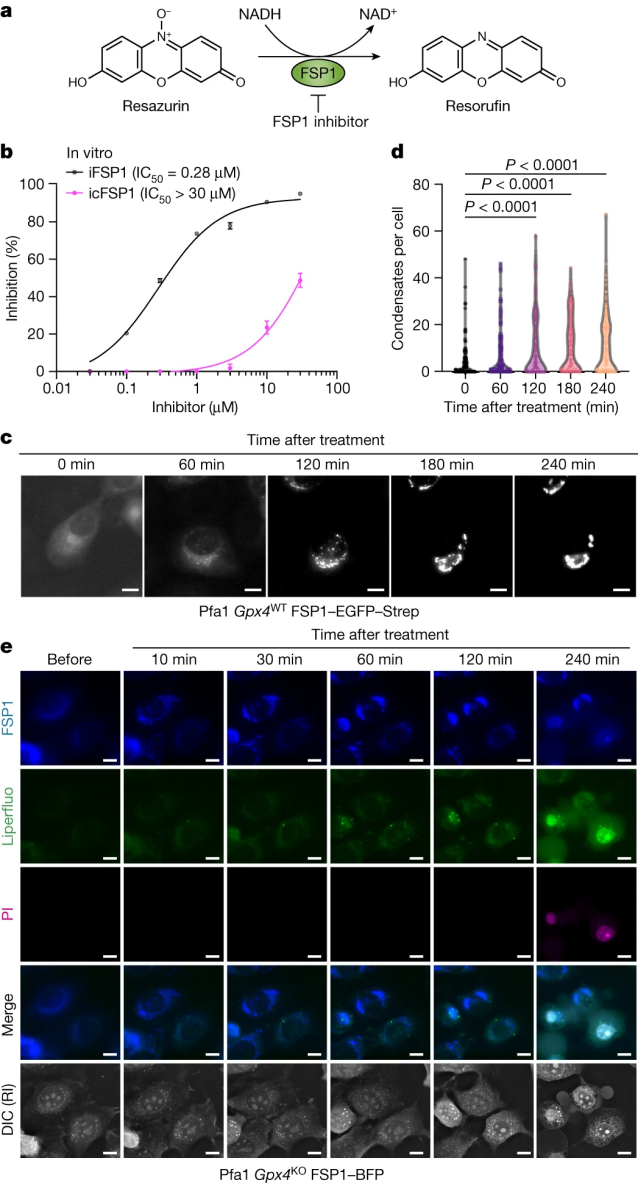
图2 icFSP1通过诱导凝聚物形成间接抑制FSP1
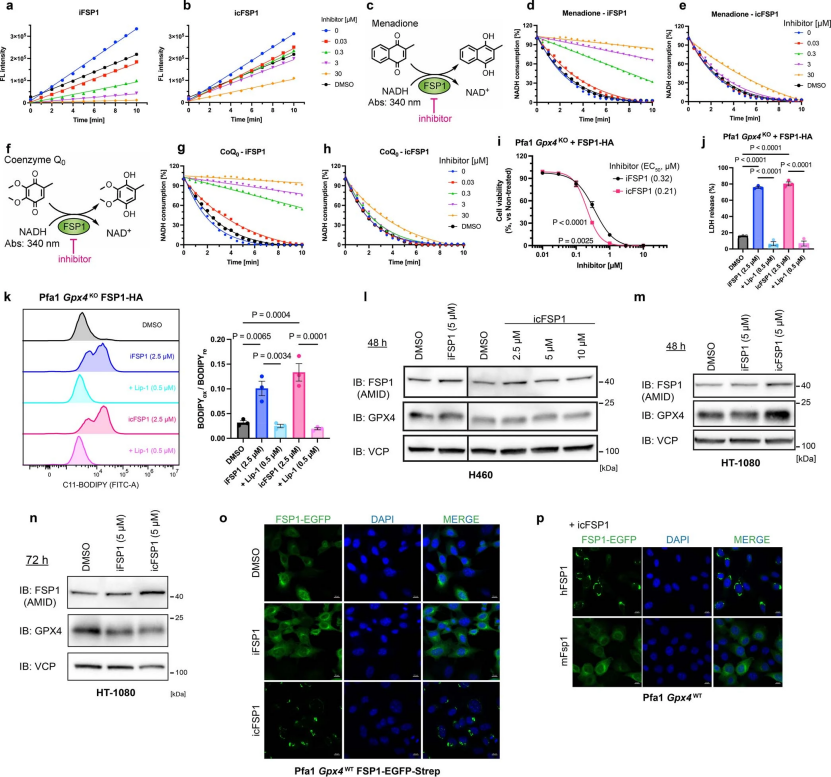
辅图3 icFSP1对FSP1酶的活性和表达无影响
3.icFSP1诱导FSP1相分离
上述实验已经证实了Nrg4缺乏会加重小鼠内皮损伤和炎症,为了探究其是否会加速动脉粥样硬化的形成,研究人员选择6周龄的AKO或apoe和nrg4双敲除(DKO)小鼠喂养WD 12周。结果显示,在DKO小鼠中,Nrg4缺失损害了血管内皮依赖性舒张作用(图2a),并增加了动脉粥样硬化病变面积(3.1倍)或主动脉根横切面(2.6倍)(图2c-f)。此外,与AKO小鼠相比,DKO小鼠血管平滑肌细胞(VSMCs)或胶原蛋白减少,巨噬细胞和T淋巴细胞浸润面积增加(图2g,h),坏死核心区占病变面积的比例增加,胶原纤维厚度减少(图2i,j),主动脉组织MMP2和MMP9的表达增加(图2k,l),MAECs中炎症因子和粘附分子的表达也有所增加(图2m)。总之,Nrg4缺失使AKO小鼠更容易发生动脉粥样硬化和动脉粥样硬化斑块不稳定。
为了探究hFSP1凝聚物是否可以定位到特定的亚细胞区室,研究人员用许多细胞器特异性标记物(小编注:例如使用EEA1标记核内体,使用LAMP1标记溶酶体,使用GM130标记高尔基体,使用Calnexin标记内质网等)对表达hFSP1-EGFP-Strep的细胞进行了共染色。据报道,FSP1定位于不同的亚细胞结构,包括内质网(ER)、高尔基体、脂滴和核周结构(辅图4a)。然而,用icFSP1处理细胞并没有诱导出明确与这些亚细胞结构共定位的hFSP1凝聚物。此外,hFSP1凝聚物也不与其他细胞器共定位,如内含体、溶酶体、线粒体、泛素依赖性聚集体或应激颗粒(G3BP1)(辅图4b,c)。研究人员在H460细胞(只表达内源性的hFSP1)中作了进一步的验证,结果显示icFSP1处理后可以检测到凝聚物的存在,但是在用多西环素诱导敲除H460细胞中的FSP1,也能显示出少量的hFSP1凝聚物(辅图4d-g)。研究人员进一步注意到,在icFSP1处理下,hFSP1凝聚物在细胞中动态、自由地移动并融合(图3a,辅图4g),在去除抑制剂后,这种自由移动的状态是可逆的(图3b)。为了更加详细地研究凝聚物的状态,研究人员建立了光漂白后荧光恢复(FRAP)分析,研究结果表明,和晚期出现的凝聚物相比(辅图5a,b),早期的凝聚物会出现FRAP现象(小编注:因为随着时间的延长,蛋白凝聚物的动态变差,凝聚物也存在的衰老行为。2020年发表在science上的“Protein condensates as aging Maxwell fluids”作者发现,随着时间的变化,线虫PGL-3蛋白光漂白后的荧光恢复时间,及两个PGL-3蛋白凝聚物融合,发生液液相分离,形成液滴的时间都明显不同:早期阶段(<0.5h),荧光恢复快速,约为1分钟,10秒形成滴液,而晚期(46h),恢复时间约为50分钟,形成时间需要十分钟。证明了多种蛋白质凝聚物的衰老行为)(图3c,d)。
基于hFSP1的这些液滴性质,研究人员认为hFSP1凝聚物可能存在相分离现象。相分离是一个以形成可逆生物分子凝聚物为特征的物理化学过程,这些凝聚物参与应激后细胞信号的调节,并与人类疾病有关,包括癌症和神经退行性疾病。相分离还参与肿瘤组织中靶蛋白的分配,并促进神经退行性疾病相关蛋白聚集体的形成。因此,研究人员为了探究hFSP1是否能在非细胞体系中产生凝聚物,利用Strep标签从HEK293T细胞中将hFSP1-EGFP-Strep免疫沉淀,从而分离出天然豆蔻酰化的hFSP1(小编注:2019年,两篇Nature背靠背发表文献,发现FSP1只有被豆蔻酰化修饰后才能够起到抗铁死亡的功能。其中,德国组发现在FSP1的N端加上标签会完全影响其抗铁死亡的能力。因此,研究人员推断FSP1的N端可能存在一些重要的修饰位点。通过预测后发现,FSP1的N端存在经典的豆蔻酰化修饰的基序,而该基序的存在说明FSP1可能与脂质双分子存在相互联系。美国组也同样通过FSP1在脂滴以及脂膜上的定位,发现了FSP1存在豆蔻酰化的修饰。而且,通过对FSP1中豆蔻酰化修饰位点的突变进一步证明了FSP1只有被豆蔻酰化修饰后才能够起到抗铁死亡的功能。本文研究人员使用将EGFP-strep加在了FSP1的C端,因此认为此时分离出的蛋白是存在豆蔻酰化的。参考文献:[1] Doll S, et al. Nature. 2019 Nov;575(7784):693-698.;[2] Bersuker K, et al. Nature. 2019 Nov;575(7784):688-692.)。对于凝聚物的形成,研究人员将聚乙二醇(PEG)用作分子聚集剂(小编注:PEG是一种亲水性非离子聚合物,通常用于生物大分子的液-液分配和沉淀,在蛋白质晶体学中被认为是产生蛋白质晶体最成功的沉淀剂),用10%PEG重建纯化的hFSP1,与免疫沉淀的EGFP-Strep对照相比,纯化的hFSP1立即形成了粘弹性物质(图3e,辅图5c,d)。为了确认单独使用icFSP1是否可以引发hFSP1凝聚,研究人员用PEG或icFSP1对纯化后的hFSP1进行了重构。然而无论icFSP1是否存在,只要有PEG的出现hFSP1就能形成凝聚物(小编注:说明在无细胞体系情况下,是hFSP1介导了凝聚物的形成)。这些凝聚物说明,在非细胞体系中,也可以像在细胞里那样形成粘弹性的物质。但这些凝聚物的具体性质(形态、分散程度等)在非细胞体系和细胞体系中可能会有所不同。为了通过相分离强化在非细胞体系中发现的hFSP1凝聚现象,研究人员使用了从大肠杆菌中纯化的无豆蔻酰化的重组hFSP1(non-myr-FSP1)(小编注:研究人员将non-myr加在了FSP1 N端,因此认为此时分离出的蛋白不存在豆蔻酰化)。同样,hFSP1可以以FSP1和PEG浓度依赖的方式形成凝聚物(辅图5e-h)。最后,研究人员通过沉降实验来研究由PEG诱导形成的hFSP1凝聚物是否会保持稳定的粘弹性状态。在PEG存在的条件下,几乎所有的hFSP1都可以得到回收(辅图5i,j),这表明hFSP1具有通过相分离诱导凝聚物形成的倾向(小编注:2012年,Michael Rosen 和 Steven McKnight 发现纯化的蛋白及核酸等能在特定的条件下发生相分离,首次证实了相分离能够通过简单的生化实验在体外重复。体外相分离实验最大的优势是可以严格控制各个组分的浓度及反应条件(如温度、pH),更加便于研究相分离现象。体外相分离实验中会经常用到一些大分子拥挤试剂,如PEG、Dextran。目前,拥挤试剂促进相分离发生的具体的机制仍不明确,一个可能的原因是拥挤试剂模拟了细胞内的高分子浓度的环境。但也有很多研究人员指出,在很多情况下,添加到实验中的大分子拥挤试剂的量可能超过存在于细胞内的环境。因此,应当慎重使用大分子拥挤试剂。如果使用大分子拥挤试剂,建议使用多种大分子拥挤试剂做实验,以排除由于大分子拥挤试剂带来的假阳性的结果。例如,有研究人员发现,可能80%左右的蛋白在有足够多大分子拥挤试剂存在的时候都可以发生相分离)。
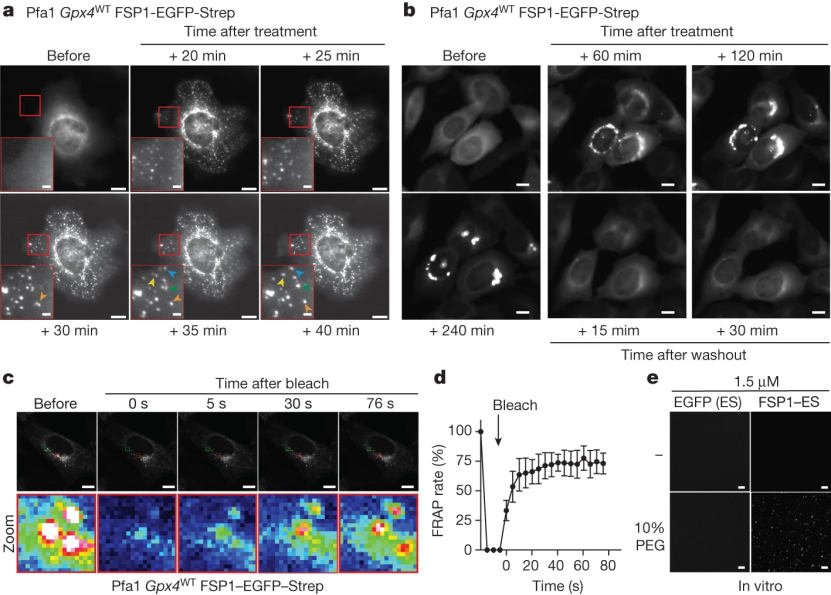
图3 FSP1凝聚物是液滴
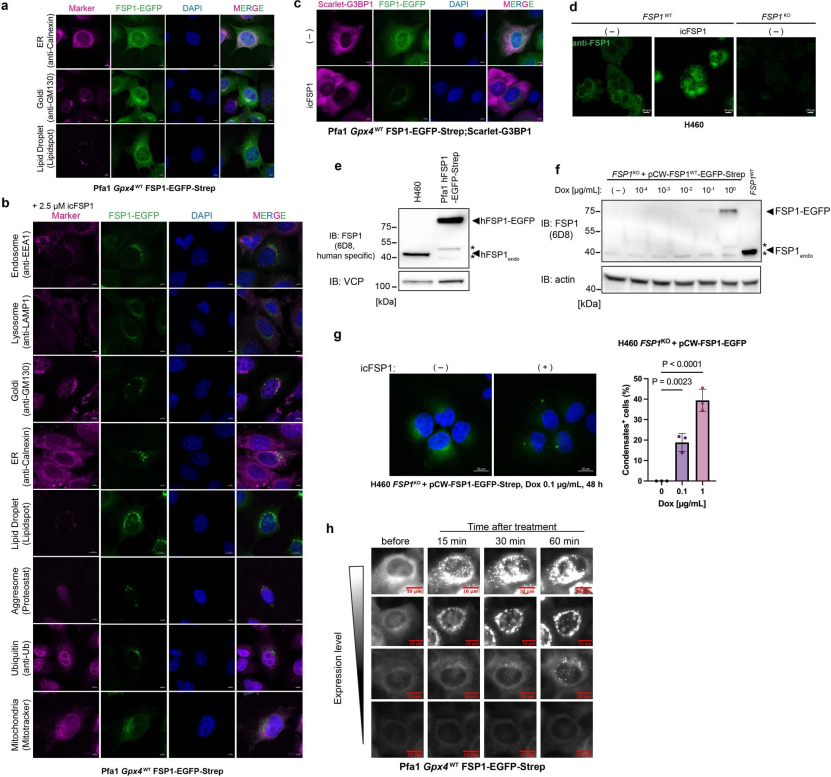
辅图4 FSP1凝聚物不定位于特定的细胞器
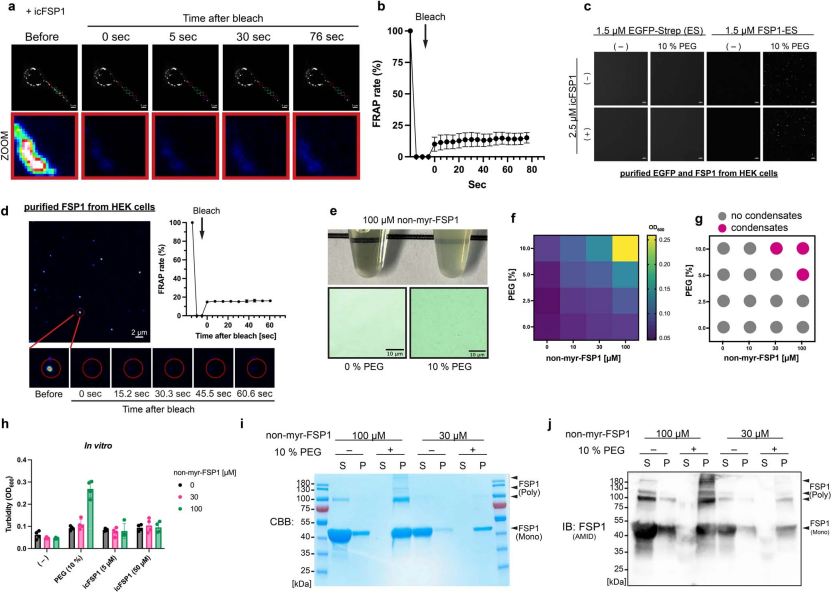
辅图5 FSP1形成粘弹性物质
拓展阅读
相分离
早在19世纪30年代,人们就描述了核仁等无膜隔室的存在,而目前越来越多的证据表明,液-液相分离(LLPS)是细胞中无膜体形成的基础。这些通过LLPS过程形成的细胞区室不断地被发现,并涉及到无数细胞功能。除了无膜体外,其他亚细胞结构也通过LLPS形成,并具有类似的潜在相互作用和物理性质,这些结构包括异染色质、核孔复合物中的运输通道,以及细胞膜上的膜受体簇。蛋白质或核酸等大分子溶液经历LLPS后会凝结成致密相,通常类似于液滴,并且这种致密相与稀相共存。而溶液是否发生相分离在很大程度上取决于大分子和溶液的浓度和特性,也取决于环境条件,包括温度、盐的类型和浓度、共溶质、pH等。
通过LLPS可以局部浓缩凝聚物中的分子来激活反应、信号传导过程和细胞骨架结构的成核。增加凝聚物中关键酶或蛋白质复合物的局部浓度可以加速生化反应。并且LLPS可以隔离分子以防止发生反应或使其失活。
参考文献
[1] Alberti, Simon et al. Cell. 176,3 (2019): 419-434.
4.液滴形成的结构基础
相分离通过分子间的多价相互作用使得细胞中无膜区室形成,通常涉及内在无序区(IDRs)和低复杂性区(LCRs)。相分离预测提示,hFSP1在其序列中包含两个假定的IDRs和一个LCR(辅图6a),这些区域可能是通过相分离形成凝聚物所必需的。为了详细分析这些预测结构域的作用,研究人员设计了以下一系列的hFSP1缺失突变体:△IDR1,△LCR,△N(△IDR1和△LCR)和△IDR2(图4a)。另外两个已知膜定位受到影响的突变体是:(1)G2A突变体,这是一个豆蔻酰基化缺陷突变体,影响其定位导致FSP1抑制铁死亡的功能消失;(2)Lyn11-G2A突变体,包含有Lyn11膜靶向序列,其N端融合到FSP1G2A上,可以抑制铁死亡。经过icFSP1处理后,只有野生型的hFSP1和Lyn11-hFSP1G2A可以改变其亚细胞定位形成凝聚物,其他突变体在icFSP1处理后均未形成hFSP1凝聚体(图4b)。与此相一致的是,用豆蔻酰化抑制剂IMP-1088预处理过的细胞会产生胞内hFSP1凝聚物消除的现象(辅图6b)。为了研究hFSP1缺失突变体是否能在非细胞体系中形成凝聚物,研究人员用10%的PEG从转染的HEK293T细胞中重组纯化的hFSP1突变体(小编注:10%的PEG是模拟生物大分子拥挤的一个环境,使用10%PEG处理之后,就更容易发生相分离)。结果显示,在PEG存在下,只有野生型hFSP1、G2A和Lyn11-G2A突变体在形成凝聚物,而IDR和LCR缺失突变体不形成凝聚物(辅图6c)。研究人员进一步从大肠杆菌中纯化具有肉豆蔻酰化的重组hFSP1(myr-FSP1)(辅图6d),并测试了豆蔻酰化hFSP1是否可以在PEG、低盐浓度和icFSP1诱导下进行体外相分离,结果显示PEG和低盐浓度似乎促进了豆蔻酰化hFSP1在体外的相分离,而icFSP1单独不能诱导相分离(辅图6e,f)。这表明细胞环境(即其他结合分子、膜环境、翻译后修饰等)对体外相分离很重要。此外,icFSP1可以诱导豆蔻酰化的hFSP1形成相分离,而即使在Pfa1细胞中存在野生型酶的hFSP1凝聚物,非豆蔻酰化的hFSP1G2A也不会形成凝聚物(辅图6g)。这些数据表明,豆蔻酰化标签的存在有助于凝聚物的形成,正如其他豆蔻酰化蛋白一样(如zeste2(EZH2))。此外,豆蔻酰化可以作为一种icFSP1的配体增强相分离的多相连接。
为了研究这些突变体潜在的铁死亡抑制功能,研究人员使用RSL3和TAM诱导的GPX4敲除细胞,进行了细胞活力测试(辅图6h,i),结果显示只有野生型的hFSP1和Lyn11-hFSP1G2A可以有效地抑制铁死亡。因此,这些数据表明,FSP1抑制铁死亡过程离不开IDRs和LCR结构域,icFSP1诱导铁死亡由hFSP1通过相分离而引发的。
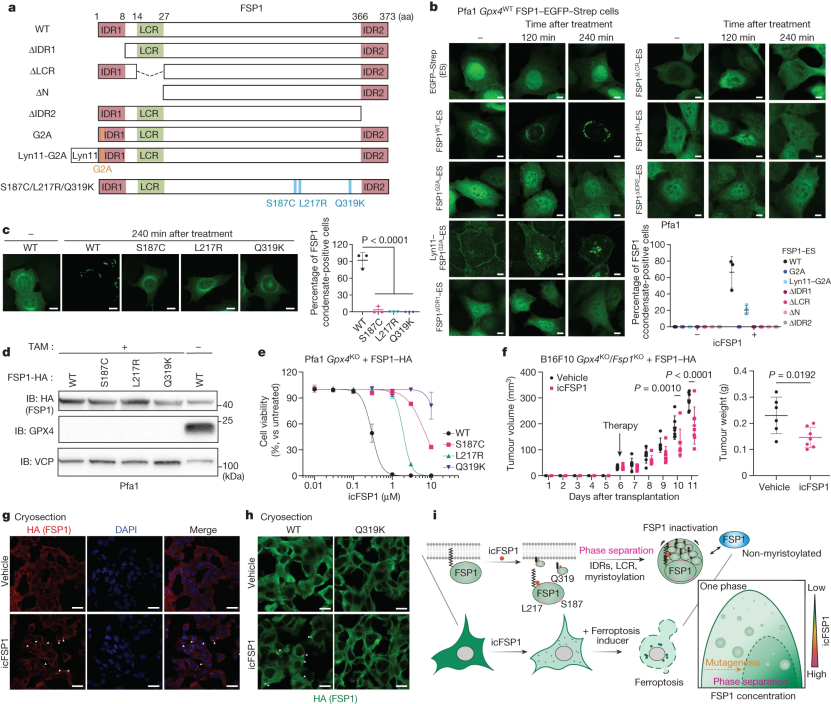
图4 FSP1的不同结构特征是相分离所必需的

辅图6 豆蔻酰化、IDRs和LCR是FSP1抗铁死亡作用和凝聚物所必需的
5.S187、L217和Q319促进了凝聚物的形成
由于icFSP1对人类酶具有特异性,并且不抑制对应鼠源的酶,因此研究人员探讨了可能解释这种物种特异性抑制活性的潜在分子机制(辅图7a)。相分离预测显示mFSP1含有两个预测的IDR和一个LCR,在其N端有一些序列差异。因此,研究人员决定构建一种嵌合酶(即hmFSP1),由hFSP1的前27个残基(包括IDR1和LCR)与mFSP1的28-373个残基融合而成。然而,这种hmFSP1嵌合酶并没有形成凝聚物(辅图7b,c),这意味着其他氨基酸可能也介导了icFSP1诱导hFSP1形成凝聚物的过程。基于人和小鼠同源基因之间的氨基酸差异,研究人员在人类FSP1中引入了一系列的点突变,并在Pfa1细胞中稳定表达了相应的突变体(辅图7d),从而使研究人员能够确定hFSP1的S187、L217和Q319残基对icFSP1依赖性凝聚物形成和铁死亡诱导至关重要,因为这些位点的替换使得突变蛋白具有抗性(图4c-e和辅图7d,e)(小编注:研究人员通过对人和小鼠的FSP1蛋白序列进行对比,筛选得到31个有差异的残基,一一对其进行点突变后筛选得到了S187C、L217R和Q319K)。鉴于(1)icFSP1与FSP1的结合并不受到这些位点替换的影响(辅图7f);(2)在这三个位点反向替换为人源残基后使mFSP1形成icFSP1依赖的凝聚物并诱导铁死亡(辅图7g,i),说明在icFSP1存在的情况下,S187,L217和Q319可能对FSP1-FSP1相互作用产生关键影响,从而触发细胞中的相分离(尽管icFSP1与野生型hFSP1确切结合位点仍有待结构解析)。

辅图7 人源FSP1对icFSP1抗性的突变分析
6.icFSP1在体内抑制肿瘤生长
为了评估icFSP1是否适用于体内使用,研究人员进行了代谢稳定性和药代动力学分析,结果表明,与iFSP1相比icFSP1明显改善了小鼠肝微粒体稳定性及其在血浆中的最大浓度(辅图8a,b)(小编注:为了初步评估化合物在体内被代谢清除情况,通常采用化合物与肝微粒体在体外共孵育一段时间,通过检测药物浓度的变化来反映化合物的稳定性)。此外,为了评估icFSP1在荷瘤小鼠模型中的作用,研究人员将过表达hFSP1的Gpx4和Fsp1双敲除的B16F10细胞注射到雌性C57BL/6J小鼠皮下。当肿瘤达到约25-50 mm3大小时,将小鼠随机分两组每天两次腹腔注射对照溶剂或icFSP1。结果显示icFSP1治疗显著抑制肿瘤生长,降低肿瘤质量,但不影响体重(图4f和辅图8c)。值得注意的是,用icFSP1对肿瘤小鼠治疗显著增加了hFSP1凝聚物的丰度和对脂质过氧化分解产物4-羟基烯醛(4-HNE)的免疫反应性(图4g和辅图8d)。这些数据表明,icFSP1可能触发hFSP1的相分离,从而抑制肿瘤在体内的生长。为了证实这些发现,研究人员建立了过表达hFSP1Q319K的Gpx4和Fsp1双敲除的B16F10突变细胞系来研究体内凝聚物的形成。与Pfa1和H460细胞获得的结果一样,依赖于hFSP1Q319K的黑色素瘤细胞系在培养细胞和体内对icFSP1具有抗性(辅图8e-j);与此相一致,在体内处理icFSP1后也未观察到FSP1凝聚物(图4h和辅图8h)。
为了研究icFSP1是否也可能在人体环境中发挥作用,研究人使用了人类黑色素瘤细胞系(A375)和人类肺癌细胞系(H460),这两种细胞系已知可以表达大量的FSP1。事实上,GPX4敲除细胞对icFSP1处理高度敏感,并且在相应的异种移植肿瘤模型中,GPX4基因敲除细胞的肿瘤生长被显著抑制(辅图9a-i)。总之,以上研究结果表明,hFSP1特异性抑制剂icFSP1可能触发FSP1的相分离,并与典型铁死亡抑制剂协同作用诱导铁死亡,以此作为一种有效的根除某些实体癌症的可行方法。
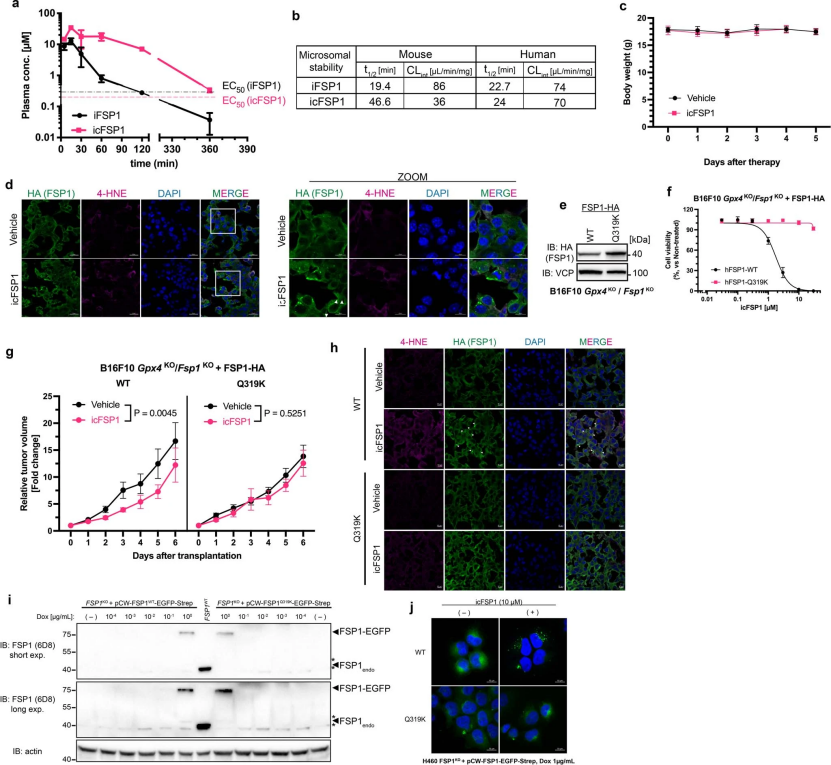
辅图8 icFSP1靶向FSP1作为潜在小鼠细胞的抗癌疗法

辅图9 icFSP1靶向FSP1作为人类细胞潜在的抗癌疗法
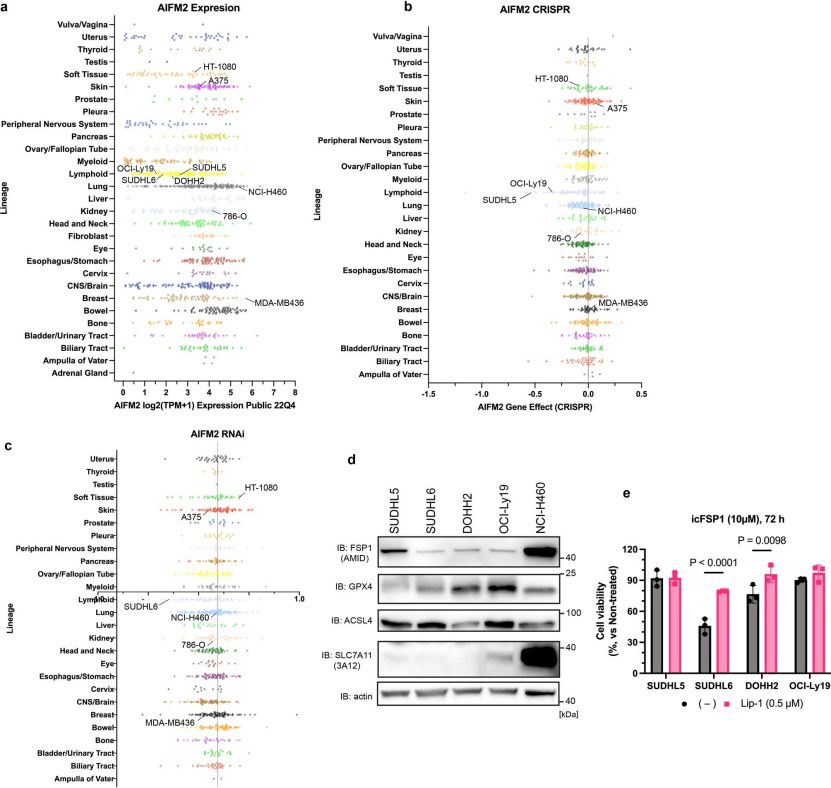
辅图10 FSP1是多种癌细胞的潜在靶点
总结
在本文中,研究人员鉴定开发了一类体内有效的新型hFSP1抑制剂——3-苯基喹唑啉酮(icFSP1),其通过触发FSP1从膜上解离并形成相分离相关的凝聚物来促进铁死亡。研究人员发现icFSP1介导hFSP1的相分离过程与hFSP1的关键分子特征:N端豆蔻酰化,特定的氨基酸残基(S187,L127和Q319),IDRs和LCR息息相关。此外,研究人员证明hFSP1特异性抑制剂icFSP1通过触发FSP1的相分离,与典型铁死亡抑制剂通过不同的机制,共同诱导肿瘤细胞的铁死亡。因此,FSP1可作为未来肿瘤治疗的有吸引力的靶标,有望通过触发铁死亡,作为一种新的抗癌方式来有效地抑制肿瘤生长并根除肿瘤。
原文链接:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10338336/
关注微信公众号代谢学人
了解更多代谢前沿资讯

https://m.sciencenet.cn/blog-3483272-1398499.html
上一篇:代谢学人Cell Metabolism:肥胖和肿瘤的幕后黑手:SNORD46
下一篇:代谢学人Nature Metabolism:产热脂肪与交感神经之“锌”的秘密